-
Atacand Tablets (Astrazeneca LP)
USE IN PREGNANCY
When used in pregnancy during the second and third trimesters, drugs that act directly on the renin-angiotensin system can cause injury and even death to the developing fetus. When pregnancy is detected, ATACAND should be discontinued as soon as poss-ible. See WARNINGS , Fetal/Neonatal Morbidity and Mortality .
DESCRIPTION
ATACAND (candesartan cilexetil), a prodrug, is hydrolyzed to candesartan during absorption from the gastrointestinal tract. Candesartan is a selective AT 1 subtype angiotensin II receptor antagonist.
Candesartan cilexetil, a nonpeptide, is chemically described as (±)-1-Hydroxyethyl 2-ethoxy-1- [ p -(o-1 H -tetrazol-5-ylphenyl)benzyl]-7-benzimidazolecarboxylate, cyclohexyl carbonate (ester).
Its empirical formula is C 33 H 34 N 6 O 6 , and its structural formula is
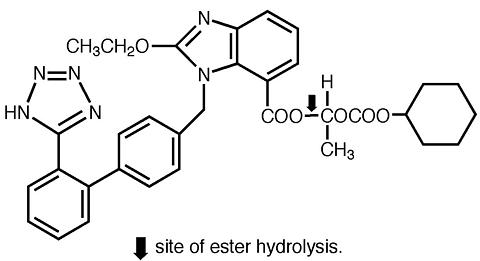
Candesartan cilexetil is a white to off-white powder with a molecular weight of 610.67. It is practically insoluble in water and sparingly soluble in methanol. Candesartan cilexetil is a racemic mixture containing one chiral center at the cyclohexyloxycarbonyloxy ethyl ester group. Following oral administration, candesartan cilexetil undergoes hydrolysis at the ester link to form the active drug, candesartan, which is achiral.
ATACAND is available for oral use as tablets containing either 4 mg, 8 mg, 16 mg, or 32 mg of candesartan cilexetil and the following inactive ingredients: hydroxypropyl cellulose, polyethylene glycol, lactose, corn starch, carboxymethylcellulose calcium, and magnesium stearate. Ferric oxide (reddish brown) is added to the 8-mg, 16-mg, and 32-mg tablets as a colorant.
CLINICAL PHARMACOLOGY
Mechanism of Action
Angiotensin II is formed from angiotensin I in a reaction catalyzed by angiotensin-converting enzyme (ACE, kininase II). Angiotensin II is the principal pressor agent of the renin-angiotensin system, with effects that include vasoconstriction, stimulation of synthesis and release of aldosterone, cardiac stimulation, and renal reabsorption of sodium. Candesartan blocks the vasoconstrictor and aldosterone-secreting effects of angiotensin II by selectively blocking the binding of angiotensin II to the AT 1 receptor in many tissues, such as vascular smooth muscle and the adrenal gland. Its action is, therefore, independent of the pathways for angiotensin II synthesis.
There is also an AT 2 receptor found in many tissues, but AT 2 is not known to be associated with cardiovascular homeostasis. Candesartan has much greater affinity (>10,000-fold) for the AT 1 receptor than for the AT 2 receptor.
Blockade of the renin-angiotensin system with ACE inhibitors, which inhibit the biosynthesis of angiotensin II from angiotensin I, is widely used in the treatment of hypertension. ACE inhibitors also inhibit the degradation of bradykinin, a reaction also catalyzed by ACE. Because candesartan does not inhibit ACE (kininase II), it does not affect the response to bradykinin. Whether this difference has clinical relevance is not yet known. Candesartan does not bind to or block other hormone receptors or ion channels known to be important in cardiovascular regulation.
Blockade of the angiotensin II receptor inhibits the negative regulatory feedback of angiotensin II on renin secretion, but the resulting increased plasma renin activity and angiotensin II circulating levels do not overcome the effect of candesartan on blood pressure.
Pharmacokinetics
General
Candesartan cilexetil is rapidly and completely bioactivated by ester hydrolysis during absorption from the gastrointestinal tract to candesartan, a selective AT 1 subtype angiotensin II receptor antagonist. Candesartan is mainly excreted unchanged in urine and feces (via bile). It undergoes minor hepatic metabolism by O-deethylation to an inactive metabolite. The elimination half-life of candesartan is approximately 9 hours. After single and repeated administration, the pharmacokinetics of candesartan are linear for oral doses up to 32 mg of candesartan cilexetil. Candesartan and its inactive metabolite do not accumulate in serum upon repeated once-daily dosing.
Following administration of candesartan cilexetil, the absolute bioavailability of candesartan was estimated to be 15%. After tablet ingestion, the peak serum concentration (C max ) is reached after 3 to 4 hours. Food with a high fat content does not affect the bioavailability of candesartan after candesartan cilexetil administration.
Metabolism and Excretion
Total plasma clearance of candesartan is 0.37 mL/min/kg, with a renal clearance of 0.19 mL/min/kg. When candesartan is administered orally, about 26% of the dose is excreted unchanged in urine. Following an oral dose of 14 C-labeled candesartan cilexetil, approximately 33% of radioactivity is recovered in urine and approximately 67% in feces. Following an intravenous dose of 14 C-labeled candesartan, approximately 59% of radioactivity is recovered in urine and approximately 36% in feces. Biliary excretion contributes to the elimination of candesartan.
Distribution
The volume of distribution of candesartan is 0.13 L/kg. Candesartan is highly bound to plasma proteins (>99%) and does not penetrate red blood cells. The protein binding is constant at candesartan plasma concentrations well above the range achieved with recommended doses. In rats, it has been demonstrated that candesartan crosses the blood-brain barrier poorly, if at all. It has also been demonstrated in rats that candesartan passes across the placental barrier and is distributed in the fetus.
Special Populations
Pediatric-- The pharmacokinetics of candesartan cilexetil have not been investigated in patients <18 years of age.
Geriatric and Gender-- The pharmacokinetics of candesartan have been studied in the elderly (>/=65 years) and in both sexes. The plasma concentration of candesartan was higher in the elderly (C max was approximately 50% higher, and AUC was approximately 80% higher) compared to younger subjects administered the same dose. The pharmacokinetics of candesartan were linear in the elderly, and candesartan and its inactive metabolite did not accumulate in the serum of these subjects upon repeated, once-daily administration. No initial dosage adjustment is necessary. (See DOSAGE AND ADMINISTRATION .) There is no difference in the pharmacokinetics of candesartan between male and female subjects.
Renal Insufficiency-- In hypertensive patients with renal insufficiency, serum concentrations of candesartan were elevated. After repeated dosing, the AUC and C max were approximately doubled in patients with severe renal impairment (creatinine clearance <30 mL/min/1.73m 2 ) compared to patients with normal kidney function. The pharmacokinetics of candesartan in hypertensive patients undergoing hemodialysis are similar to those in hypertensive patients with severe renal impairment. Candesartan cannot be removed by hemodialysis. No initial dosage adjustment is necessary in patients with renal insufficiency. (See DOSAGE AND ADMINISTRATION .)
In heart failure patients with renal impairment, AUC 0-72h was 36% and 65% higher in mild and moderate renal impairment, respectively. C max was 15% and 55% higher in mild and moderate renal impairment, respectively.
Hepatic Insufficiency-- The pharmacokinetics of candesartan were compared in patients with mild and moderate hepatic impairment to matched healthy volunteers following a single oral dose of 16 mg candesartan cilexetil. The increase in AUC for candesartan was 30% in patients with mild hepatic impairment (Child-Pugh A) and 145% in patients with moderate hepatic impairment (Child-Pugh B). The increase in C max for candesartan was 56% in patients with mild hepatic impairment and 73% in patients with moderate hepatic impairment. The pharmacokinetics after candesartan cilexetil administration have not been investigated in patients with severe hepatic impairment. No initial dosage adjustment is necessary in patients with mild hepatic impairment. In hypertensive patients with moderate hepatic impairment, consideration should be given to initiation of ATACAND at a lower dose. (See DOSAGE AND ADMINISTRATION .)
Heart Failure-- The pharmacokinetics of candesartan were linear in patients with heart failure (NYHA class II and III) after candesartan cilexetil doses of 4, 8, and 16 mg. After repeated dosing, the AUC was approximately doubled in these patients compared with healthy, younger patients. The pharmacokinetics in heart failure patients is similar to that in healthy elderly volunteers. (See DOSAGE AND ADMINISTRATION , Heart Failure .)
Drug Interactions
See PRECAUTIONS , Drug Interactions .
Pharmacodynamics
Candesartan inhibits the pressor effects of angiotensin II infusion in a dose-dependent manner. After 1 week of once daily dosing with 8 mg of candesartan cilexetil, the pressor effect was inhibited by approximately 90% at peak with approximately 50% inhibition persisting for 24 hours.
Plasma concentrations of angiotensin I and angiotensin II, and plasma renin activity (PRA), increased in a dose-dependent manner after single and repeated administration of candesartan cilexetil to healthy subjects, hypertensive, and heart failure patients. ACE activity was not altered in healthy subjects after repeated candesartan cilexetil administration. The once-daily administration of up to 16 mg of candesartan cilexetil to healthy subjects did not influence plasma aldosterone concentrations, but a decrease in the plasma concentration of aldosterone was observed when 32 mg of candesartan cilexetil was administered to hypertensive patients. In spite of the effect of candesartan cilexetil on aldosterone secretion, very little effect on serum potassium was observed.
Hypertension
In multiple-dose studies with hypertensive patients, there were no clinically significant changes in metabolic function, including serum levels of total cholesterol, triglycerides, glucose, or uric acid. In a 12-week study of 161 patients with non-insulin-dependent (type 2) diabetes mellitus and hypertension, there was no change in the level of HbA 1c .
Heart Failure
In heart failure patients, candesartan >/=8 mg resulted in decreases in systemic vascular resistance and pulmonary capillary wedge pressure.
Clinical Trials
Hypertension
The antihypertensive effects of ATACAND were examined in 14 placebo-controlled trials of 4- to 12-weeks duration, primarily at daily doses of 2 to 32 mg per day in patients with baseline diastolic blood pressures of 95 to 114 mm Hg. Most of the trials were of candesartan cilexetil as a single agent, but it was also studied as add-on to hydrochlorothiazide and amlodipine. These studies included a total of 2350 patients randomized to one of several doses of candesartan cilexetil and 1027 to placebo. Except for a study in diabetics, all studies showed significant effects, generally dose related, of 2 to 32 mg on trough (24 hour) systolic and diastolic pressures compared to placebo, with doses of 8 to 32 mg giving effects of about 8-12/4-8 mm Hg. There were no exaggerated first-dose effects in these patients. Most of the antihypertensive effect was seen within 2 weeks of initial dosing, and the full effect in 4 weeks. With once-daily dosing, blood pressure effect was maintained over 24 hours, with trough to peak ratios of blood pressure effect generally over 80%. Candesartan cilexetil had an additional blood pressure lowering effect when added to hydrochlorothiazide.
The antihypertensive effects of candesartan cilexetil and losartan potassium at their highest recommended doses administered once-daily were compared in two randomized, double-blind trials. In a total of 1268 patients with mild to moderate hypertension who were not receiving other antihypertensive therapy, candesartan cilexetil 32 mg lowered systolic and diastolic blood pressure by 2 to 3 mm Hg on average more than losartan potassium 100 mg, when measured at the time of either peak or trough effect. The antihypertensive effects of twice daily dosing of either candesartan cilexetil or losartan potassium were not studied.
The antihypertensive effect was similar in men and women and in patients older and younger than 65. Candesartan was effective in reducing blood pressure regardless of race, although the effect was somewhat less in blacks (usually a low-renin population). This has been generally true for angiotensin II antagonists and ACE inhibitors.
In long-term studies of up to 1 year, the antihypertensive effectiveness of candesartan cilexetil was maintained, and there was no rebound after abrupt withdrawal.
There were no changes in the heart rate of patients treated with candesartan cilexetil in controlled trials.
Heart Failure
Candesartan was studied in two heart failure outcome studies: 1. The Candesartan in Heart failure: Assessment of Reduction in Mortality and morbidity trial in patients intolerant of ACE inhibitors (CHARM- Alternative), 2. CHARM-Added in patients already receiving ACE inhibitors. Both studies were international double-blind, placebo-controlled trials in patients with NYHA class II - IV heart failure and LVEF </=40%. In both trials, patients were randomized to placebo or ATACAND (initially 4-8 mg once daily, titrated as tolerated to 32 mg once daily) and followed for up to 4 years. Patients with serum creatinine >/=3 mg/dL, serum potassium >/=5.5 mEq/L, symptomatic hypotension or known bilateral renal artery stenosis were excluded. The primary end point in both trials was time to either cardiovascular death or hospitalization for heart failure.
CHARM-Alternative included 2028 subjects not receiving an ACE inhibitor due to intolerance. The mean age was 67 years and 32% were female, 48% were NYHA II, 49% were NYHA III, 4% were NYHA IV, and the mean ejection fraction was 30%. Sixty-two percent had a history of myocardial infarction, 50% had a history of hypertension, and 27% had diabetes. Concomitant drugs at baseline were diuretics (85%), digoxin (46%), beta-blockers (55%), and spironolactone (24%). The mean daily dose of ATACAND was approximately 23 mg and 59% of subjects on treatment received 32 mg once daily.
After a median follow-up of 34 months, there was a 23% reduction in the risk of cardiovascular death or heart failure hospitalization on ATACAND (p<0.001), with both components contributing to the overall effect (Table 1).
Table 1. CHARM - Alternative: Primary Endpoint and its Components Endpoint (time to first event)ATACAND
(n=1013)Placebo
(n=1015)Hazard Ratio
(95% CI)p-value
(logrank)CV death or heart failure hospitalization334 406 0.77
(0.67-0.89)<0.001 CV death219 252 0.85
(0.71-1.02)0.072 Heart failure hospitalization207 286 0.68
(0.57-0.81)<0.001 In CHARM-Added, 2548 subjects receiving an ACE inhibitor were randomized to ATACAND or placebo. The specific ACE inhibitor and dose were at the discretion of the investigators, who were encouraged to titrate patients to doses known to be effective in clinical outcome trials, subject to patient tolerability. Forced titration to maximum tolerated doses of ACE inhibitor was not required.
The mean age was 64 years and 21% were female, 24% were NYHA II, 73% were NYHA III, 3% were NYHA IV, and the mean ejection fraction was 28%. Fifty-six percent had a history of myocardial infarction, 48% had a history of hypertension, and 30% had diabetes. Concomitant drugs at baseline in addition to ACE inhibitors were diuretics (90%), digoxin (58%), beta-blockers (55%), and spironolactone (17%). The mean daily dose of ATACAND was approximately 24 mg and 61% of subjects on treatment received 32 mg once daily.
After a median follow-up of 41 months, there was a 15% reduction in the risk of cardiovascular death or heart failure hospitalization on ATACAND (p=0.011), with both components contributing to the overall effect (Table 2). There was no evident relationship between dose of ACE inhibitor and the benefit of ATACAND.
Table 2. CHARM - Added: Primary Endpoint and its Components Endpoint (time to first event)ATACAND
(n=1276)Placebo
(n=1272)Hazard Ratio
(95% CI)p-value
(logrank)CV death or heart failure hospitalization483 538 0.85
(0.75-0.96)0.011 CV death302 347 0.84
(0.72-0.98)0.029 Heart failure hospitalization309 356 0.83
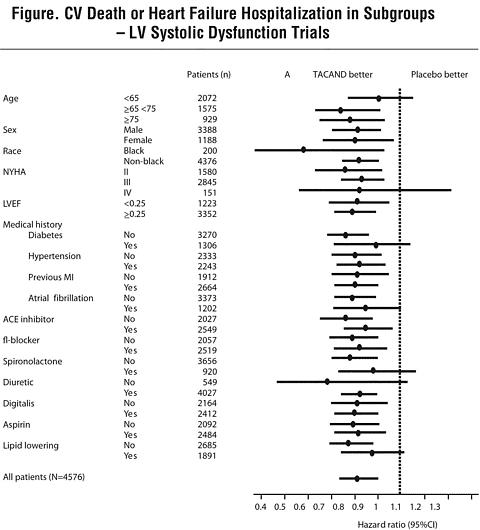
(0.71-0.96)0.014 In these two studies, the benefit of ATACAND in reducing the risk of CV death or heart failure hospitalization (18% p<0.001) was evident in major subgroups (see Figure), and in patients on other combinations of cardiovascular and heart failure treatments, including ACE inhibitors and beta-blockers.
INDICATIONS AND USAGE
Hypertension
ATACAND is indicated for the treatment of hypertension. It may be used alone or in combination with other antihypertensive agents.
Heart Failure
ATACAND is indicated for the treatment of heart failure (NYHA class II-IV) in patients with left ventricular systolic dysfunction (ejection fraction </=40%) to reduce cardiovascular death and to reduce heart failure hospitalizations. (See Clinical Trials .) ATACAND also has an added effect on these outcomes when used with an ACE inhibitor.
CONTRAINDICATIONS
ATACAND is contraindicated in patients who are hypersensitive to any component of this product.
WARNINGS
Fetal/Neonatal Morbidity and Mortality
Drugs that act directly on the renin-angiotensin system can cause fetal and neonatal morbidity and death when administered to pregnant women. Several dozen cases have been reported in the world literature in patients who were taking angiotensin-converting enzyme inhibitors. Post-marketing experience has identified reports of fetal and neonatal toxicity in babies born to women treated with ATACAND during pregnancy. When pregnancy is detected, ATACAND should be discontinued as soon as possible.
The use of drugs that act directly on the renin-angiotensin system during the second and third trimesters of pregnancy has been associated with fetal and neonatal injury, including hypotension, neonatal skull hypoplasia, anuria, reversible or irreversible renal failure, and death. Oligohydramnios has also been reported, presumably resulting from decreased fetal renal function; oligohydramnios in this setting has been associated with fetal limb contractures, craniofacial deformation, and hypoplastic lung development. Prematurity, intrauterine growth retardation, and patent ductus arteriosus have also been reported, although it is not clear whether these occurrences were due to exposure to the drug.
These adverse effects do not appear to have resulted from intrauterine drug exposure that has been limited to the first trimester. Mothers whose embryos and fetuses are exposed to an angiotensin II receptor antagonist only during the first trimester should be so informed. Nonetheless, when patients become pregnant, physicians should have the patient discontinue the use of ATACAND as soon as possible.
Rarely (probably less often than once in every thousand pregnancies), no alternative to a drug acting on the renin-angiotensin system will be found. In these rare cases, the mothers should be apprised of the potential hazards to their fetuses, and serial ultrasound examinations should be performed to assess the intra-amniotic environment.
If oligohydramnios is observed, ATACAND should be discontinued unless it is considered life saving for the mother. Contraction stress testing (CST), a nonstress test (NST), or biophysical profiling (BPP) may be appropriate, depending upon the week of pregnancy. Patients and physicians should be aware, however, that oligohydramnios may not appear until after the fetus has sustained irreversible injury.
Infants with histories of in utero exposure to an angiotensin II receptor antagonist should be closely observed for hypotension, oliguria, and hyperkalemia. If oliguria occurs, attention should be directed toward support of blood pressure and renal perfusion. Exchange transfusion or dialysis may be required as means of reversing hypotension and/or substituting for disordered renal function.
Oral doses >/=10 mg of candesartan cilexetil/kg/day administered to pregnant rats during late gestation and continued through lactation were associated with reduced survival and an increased incidence of hydronephrosis in the offspring. The 10-mg/kg/day dose in rats is approximately 2.8 times the maximum recommended daily human dose (MRHD) of 32 mg on a mg/m 2 basis (comparison assumes human body weight of 50 kg). Candesartan cilexetil given to pregnant rabbits at an oral dose of 3 mg/kg/day (approximately 1.7 times the MRHD on a mg/m 2 basis) caused maternal toxicity (decreased body weight and death) but, in surviving dams, had no adverse effects on fetal survival, fetal weight, or external, visceral, or skeletal development. No maternal toxicity or adverse effects on fetal development were observed when oral doses up to 1000 mg of candesartan cilexetil/kg/day (approximately 138 times the MRHD on a mg/m 2 basis) were administered to pregnant mice.
Hypotension in Volume- and Salt-Depleted Patients
In patients with an activated renin-angiotensin system, such as volume- and/or salt-depleted patients (eg, those being treated with diuretics), symptomatic hypotension may occur. These conditions should be corrected prior to administration of ATACAND, or the treatment should start under close medical supervision (see DOSAGE AND ADMINISTRATION ).
If hypotension occurs, the patients should be placed in the supine position and, if necessary, given an intravenous infusion of normal saline. A transient hypotensive response is not a contraindication to further treatment which usually can be continued without difficulty once the blood pressure has stabilized.
Hypotension in Heart Failure Patients
Caution should be observed when initiating therapy in patients with heart failure. Patients with heart failure given ATACAND commonly have some reduction in blood pressure. In patients with symptomatic hypotension this may require temporarily reducing the dose of ATACAND, or diuretic, or both, and volume repletion. In the CHARM program, hypotension was reported in 18.8% of patients on candesartan versus 9.8% of patients on placebo. The incidence of hypotension leading to drug discontinuation in candesartan-treated patients was 4.1% compared with 2.0% in placebo-treated patients. Monitoring of blood pressure is recommended during dose escalation and periodically thereafter.
PRECAUTIONS
General
Impaired Hepatic Function-- Based on pharmacokinetic data which demonstrate significant increases in candesartan AUC and C max in patients with moderate hepatic impairment, a lower initiating dose should be considered for patients with moderate hepatic impairment. (See DOSAGE AND ADMINISTRATION , and CLINICAL PHARMACOLOGY , Special Populations .)
Impaired Renal Function-- As a consequence of inhibiting the renin-angiotensin-aldosterone system, changes in renal function may be anticipated in susceptible individuals treated with ATACAND. In patients whose renal function may depend upon the activity of the renin-angiotensin-aldosterone system (eg, patients with severe heart failure), treatment with angiotensin-converting enzyme inhibitors and angiotensin receptor antagonists has been associated with oliguria and/or progressive azotemia and (rarely) with acute renal failure and/or death. Similar results may be anticipated in patients treated with ATACAND. (See CLINICAL PHARMACOLOGY , Special Populations .)
In studies of ACE inhibitors in patients with unilateral or bilateral renal artery stenosis, increases in serum creatinine or blood urea nitrogen (BUN) have been reported. There has been no long-term use of ATACAND in patients with unilateral or bilateral renal artery stenosis, but similar results may be expected.
In heart failure patients treated with ATACAND, increases in serum creatinine may occur. Dosage reduction or discontinuation of the diuretic or ATACAND, and volume repletion may be required. In the CHARM program, the incidence of abnormal renal function (e.g., creatinine increase) was 12.5% in patients treated with candesartan versus 6.3% in patients treated with placebo. The incidence of abnormal renal function (eg, creatinine increase) leading to drug discontinuation in candesartan-treated patients was 6.3% compared with 2.9% in placebo-treated patients. Evaluation of patients with heart failure should always include assessment of renal function and volume status. Monitoring of serum creatinine is recommended during dose escalation and periodically thereafter.
Hyperkalemia
In heart failure patients treated with ATACAND, hyperkalemia may occur, especially when taken concomitantly with ACE inhibitors and potassium-sparing diuretics such as spironolactone. In the CHARM program, the incidence of hyperkalemia was 6.3% in patients treated with candesartan versus 2.1% in patients treated with placebo. The incidence of hyperkalemia leading to drug discontinuation in candesartan-treated patients was 2.4% compared with 0.6% in placebo-treated patients. During treatment with ATACAND in patients with heart failure, monitoring of serum potassium is recommended during dose escalation and periodically thereafter.
Information for Patients
Pregnancy-- Female patients of childbearing age should be told about the consequences of second- and third-trimester exposure to drugs that act on the renin-angiotensin system, and they should also be told that these consequences do not appear to have resulted from intrauterine drug exposure that has been limited to the first trimester. These patients should be asked to report pregnancies to their physicians as soon as possible.
Drug Interactions
No significant drug interactions have been reported in studies of candesartan cilexetil given with other drugs such as glyburide, nifedipine, digoxin, warfarin, hydrochlorothiazide, and oral contraceptives in healthy volunteers, or given with enalapril to patients with heart failure (NYHA class II and III). Because candesartan is not significantly metabolized by the cytochrome P450 system and at therapeutic concentrations has no effects on P450 enzymes, interactions with drugs that inhibit or are metabolized by those enzymes would not be expected.
Lithium-- Reversible increases in serum lithium concentrations and toxicity have been reported during concomitant administration of lithium with ACE inhibitors, and with some angiotensin II receptor antagonists. An increase in serum lithium concentration has been reported during concomitant administration of lithium with ATACAND, so careful monitoring of serum lithium levels is recommended during concomitant use.
Carcinogenesis, Mutagenesis, Impairment of Fertility
There was no evidence of carcinogenicity when candesartan cilexetil was orally administered to mice and rats for up to 104 weeks at doses up to 100 and 1000 mg/kg/day, respectively. Rats received the drug by gavage, whereas mice received the drug by dietary administration. These (maximally-tolerated) doses of candesartan cilexetil provided systemic exposures to candesartan (AUCs) that were, in mice, approximately 7 times and, in rats, more than 70 times the exposure in man at the maximum recommended daily human dose (32 mg).
Candesartan and its O-deethyl metabolite tested positive for genotoxicity in the in vitro Chinese hamster lung (CHL) chromosomal aberration assay. Neither compound tested positive in the Ames microbial mutagenesis assay or the in vitro mouse lymphoma cell assay. Candesartan (but not its O-deethyl metabolite) was also evaluated in vivo in the mouse micronucleus test and in vitro in the Chinese hamster ovary (CHO) gene mutation assay, in both cases with negative results. Candesartan cilexetil was evaluated in the Ames test, the in vitro mouse lymphoma cell and rat hepatocyte unscheduled DNA synthesis assays and the in vivo mouse micronucleus test, in each case with negative results. Candesartan cilexetil was not evaluated in the CHL chromosomal aberration or CHO gene mutation assay.
Fertility and reproductive performance were not affected in studies with male and female rats given oral doses of up to 300 mg/kg/day (83 times the maximum daily human dose of 32 mg on a body surface area basis).
Pregnancy
Pregnancy Categories C (first trimester) and D (second and third trimesters)--See WARNINGS , Fetal/Neonatal Morbidity and Mortality .
Nursing Mothers
It is not known whether candesartan is excreted in human milk, but candesartan has been shown to be present in rat milk. Because of the potential for adverse effects on the nursing infant, a decision should be made whether to discontinue nursing or discontinue the drug, taking into account the importance of the drug to the mother.
Pediatric Use
Safety and effectiveness in pediatric patients have not been established.
Geriatric Use
Hypertension
Of the total number of subjects in clinical studies of ATACAND, 21% (683/3260) were 65 and over, while 3% (87/3260) were 75 and over. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out. In a placebo-controlled trial of about 200 elderly hypertensive patients (ages 65 to 87 years), administration of candesartan cilexetil was well tolerated and lowered blood pressure by about 12/6 mm Hg more than placebo.
Heart Failure
Of the 7599 patients with heart failure in the CHARM program, 4343 (57%) were age 65 years or older and 1736 (23%) were 75 years or older. In patients >/=75 years of age, the incidence of drug discontinuations due to adverse events was higher for those treated with ATACAND or placebo compared with patients <75 years of age. In these patients, the most common adverse events leading to drug discontinuation at an incidence of at least 3%, and more frequent with ATACAND than placebo, were abnormal renal function (7.9% vs. 4.0%), hypotension (5.2% vs. 3.2%) and hyperkalemia (4.2% vs. 0.9%). In addition to monitoring of serum creatinine, potassium, and blood pressure during dose escalation and periodically thereafter, greater sensitivity of some older individuals with heart failure must be considered.
ADVERSE REACTIONS
Hypertension
ATACAND has been evaluated for safety in more than 3600 patients/subjects, including more than 3200 patients treated for hypertension. About 600 of these patients were studied for at least 6 months and about 200 for at least 1 year. In general, treatment with ATACAND was well tolerated. The overall incidence of adverse events reported with ATACAND was similar to placebo.
The rate of withdrawals due to adverse events in all trials in patients (7510 total) was 3.3% (ie, 108 of 3260) of patients treated with candesartan cilexetil as monotherapy and 3.5% (ie, 39 of 1106) of patients treated with placebo. In placebo-controlled trials, discontinuation of therapy due to clinical adverse events occurred in 2.4% (ie, 57 of 2350) of patients treated with ATACAND and 3.4% (ie, 35 of 1027) of patients treated with placebo.
The most common reasons for discontinuation of therapy with ATACAND were headache (0.6%) and dizziness (0.3%).
The adverse events that occurred in placebo-controlled clinical trials in at least 1% of patients treated with ATACAND and at a higher incidence in candesartan cilexetil (n=2350) than placebo (n=1027) patients included back pain (3% vs. 2%), dizziness (4% vs. 3%), upper respiratory tract infection (6% vs. 4%), pharyngitis (2% vs. 1%), and rhinitis (2% vs. 1%).
The following adverse events occurred in placebo-controlled clinical trials at a more than 1% rate but at about the same or greater incidence in patients receiving placebo compared to candesartan cilexetil: fatigue, peripheral edema, chest pain, headache, bronchitis, coughing, sinusitis, nausea, abdominal pain, diarrhea, vomiting, arthralgia, albuminuria.
Other potentially important adverse events that have been reported, whether or not attributed to treatment, with an incidence of 0.5% or greater from the 3260 patients worldwide treated in clinical trials with ATACAND are listed below. It cannot be determined whether these events were causally related to ATACAND. Body as a Whole: asthenia, fever; Central and Peripheral Nervous System: paresthesia, vertigo; Gastrointestinal System Disorder: dyspepsia, gastroenteritis; Heart Rate and Rhythm Disorders: tachycardia, palpitation; Metabolic and Nutritional Disorders: creatine phosphokinase increased, hyperglycemia, hypertriglyceridemia, hyperuricemia; Musculoskeletal System Disorders: myalgia; Platelet/Bleeding-Clotting Disorders: epistaxis; Psychiatric Disorders: anxiety, depression, somnolence; Respiratory System Disorders: dyspnea; Skin and Appendages Disorders: rash, sweating increased; Urinary System Disorders: hematuria.
Other reported events seen less frequently included angina pectoris, myocardial infarction, and angioedema.
Adverse events occurred at about the same rates in men and women, older and younger patients, and black and non-black patients.
Heart Failure
The adverse event profile of ATACAND in heart failure patients was consistent with the pharmacology of the drug and the health status of the patients. In the CHARM program, comparing ATACAND in total daily doses up to 32 mg once daily (n=3803) with placebo (n=3796), 21.0% of patients discontinued ATACAND for adverse events vs. 16.1% of placebo patients.
Post-Marketing Experience:
The following have been very rarely reported in post-marketing experience:
Digestive: Abnormal hepatic function and hepatitis.
Hematologic: Neutropenia, leukopenia, and agranulocytosis.
Metabolic and Nutritional Disorders: hyperkalemia, hyponatremia.
Renal: renal impairment, renal failure.
Skin and Appendages Disorders: Pruritus and urticaria.
Rare reports of rhabdomyolysis have been reported in patients receiving angiotensin II receptor blockers.
Laboratory Test Findings
Hypertension
In controlled clinical trials, clinically important changes in standard laboratory parameters were rarely associated with the administration of ATACAND.
Creatinine, Blood Urea Nitrogen -- Minor increases in blood urea nitrogen (BUN) and serum creatinine were observed infrequently.
Hyperuricemia -- Hyperuricemia was rarely found (19 or 0.6% of 3260 patients treated with candesartan cilexetil and 5 or 0.5% of 1106 patients treated with placebo).
Hemoglobin and Hematocrit -- Small decreases in hemoglobin and hematocrit (mean decreases of approximately 0.2 grams/dL and 0.5 volume percent, respectively) were observed in patients treated with ATACAND alone but were rarely of clinical importance. Anemia, leukopenia, and thrombocytopenia were associated with withdrawal of one patient each from clinical trials.
Potassium -- A small increase (mean increase of 0.1 mEq/L) was observed in patients treated with ATACAND alone but was rarely of clinical importance. One patient from a congestive heart failure trial was withdrawn for hyperkalemia (serum potassium = 7.5 mEq/L). This patient was also receiving spironolactone.
Liver Function Tests -- Elevations of liver enzymes and/or serum bilirubin were observed infrequently. Five patients assigned to candesartan cilexetil in clinical trials were withdrawn because of abnormal liver chemistries. All had elevated transaminases. Two had mildly elevated total bilirubin, but one of these patients was diagnosed with Hepatitis A.
Heart Failure
In the CHARM program, small increases in serum creatinine (mean increase 0.2 mg/dL in candesartan-treated patients and 0.1 mg/dL in placebo-treated patients) and serum potassium (mean increase 0.15 mEq/L in candesartan-treated patients and 0.02 mEq/L in placebo-treated patients), and small decreases in hemoglobin (mean decrease 0.5 gm/dL in candesartan-treated patients and 0.3 gm/dL in placebo-treated patients) and hematocrit (mean decrease 1.6% in candesartan-treated patients and 0.9% in placebo-treated patients) were observed.
OVERDOSAGE
No lethality was observed in acute toxicity studies in mice, rats, and dogs given single oral doses of up to 2000 mg/kg of candesartan cilexetil. In mice given single oral doses of the primary metabolite, candesartan, the minimum lethal dose was greater than 1000 mg/kg but less than 2000 mg/kg.
The most likely manifestation of overdosage with ATACAND would be hypotension, dizziness, and tachycardia; bradycardia could occur from parasympathetic (vagal) stimulation. If symptomatic hypotension should occur, supportive treatment should be instituted.
Candesartan cannot be removed by hemodialysis.
Treatment: To obtain up-to-date information about the treatment of overdose, consult your Regional Poison Control Center. Telephone numbers of certified poison control centers are listed in the Physicians' Desk Reference (PDR) . In managing overdose, consider the possibilities of multiple-drug overdoses, drug-drug interactions, and altered pharmacokinetics in your patient.
DOSAGE AND ADMINISTRATION
Hypertension
Dosage must be individualized. Blood pressure response is dose related over the range of 2 to 32 mg. The usual recommended starting dose of ATACAND is 16 mg once daily when it is used as monotherapy in patients who are not volume depleted. ATACAND can be administered once or twice daily with total daily doses ranging from 8 mg to 32 mg. Larger doses do not appear to have a greater effect, and there is relatively little experience with such doses. Most of the antihypertensive effect is present within 2 weeks, and maximal blood pressure reduction is generally obtained within 4 to 6 weeks of treatment with ATACAND.
No initial dosage adjustment is necessary for elderly patients, for patients with mildly impaired renal function, or for patients with mildly impaired hepatic function (see CLINICAL PHARMACOLOGY , Special Populations ). In patients with moderate hepatic impairment, consideration should be given to initiation of ATACAND at a lower dose (See CLINICAL PHARMACOLOGY , Special Populations ). For patients with possible depletion of intravascular volume (eg, patients treated with diuretics, particularly those with impaired renal function), ATACAND should be initiated under close medical supervision and consideration should be given to administration of a lower dose (see WARNINGS , Hypotension in Volume- and Salt-Depleted Patients ).
ATACAND may be administered with or without food.
If blood pressure is not controlled by ATACAND alone, a diuretic may be added. ATACAND may be administered with other antihypertensive agents.
Heart Failure
The recommended initial dose for treating heart failure is 4 mg once daily. The target dose is 32 mg once daily, which is achieved by doubling the dose at approximately 2-week intervals, as tolerated by the patient.
HOW SUPPLIED
No. 3782 -- Tablets ATACAND, 4 mg, are white to off-white, circular/biconvex-shaped, non-film-coated tablets, coded ACF on one side and 004 on the other. They are supplied as follows:
NDC 0186-0004-31 unit of use bottles of 30.
No. 3780 -- Tablets ATACAND, 8 mg, are light pink, circular/biconvex-shaped, non-film-coated tablets, coded ACG on one side and 008 on the other. They are supplied as follows:
NDC 0186-0008-31 unit of use bottles of 30.
No. 3781 -- Tablets ATACAND, 16 mg, are pink, circular/biconvex-shaped, non-film-coated tablets, coded ACH on one side and 016 on the other. They are supplied as follows:
NDC 0186-0016-31 unit of use bottles of 30
NDC 0186-0016-54 unit of use bottles of 90
NDC 0186-0016-28 unit dose packages of 100.
No. 3791 -- Tablets ATACAND, 32 mg, are pink, circular/biconvex-shaped, non-film-coated tablets, coded ACL on one side and 032 on the other. They are supplied as follows:
NDC 0186-0032-31 unit of use bottles of 30
NDC 0186-0032-54 unit of use bottles of 90
NDC 0186-0032-28 unit dose packages of 100.
Storage
Store at 25°C (77°F); excursions permitted to 15-30°C (59-86°F) [see USP Controlled Room Temperature]. Keep container tightly closed.
ATACAND is a trademark of the Astrazeneca group of companies
© Astrazeneca 2005
Manufactured under the license from
Takeda Pharmaceutical Company, Ltd.
by: Astrazeneca AB, S-151 85 Södertälje, Sweden
for: Astrazeneca LP, Wilmington, DE 19850
Made in Sweden
Rev. 05/05 Astrazeneca
Subscribe to the "News" RSS Feed 
TOP ۞