-
Avandia Tablets (Glaxosmithkline)
DESCRIPTION
AVANDIA (rosiglitazone maleate) is an oral antidiabetic agent which acts primarily by increasing insulin sensitivity. AVANDIA is used in the management of type 2 diabetes mellitus (also known as non-insulin-dependent diabetes mellitus [NIDDM] or adult-onset diabetes). AVANDIA improves glycemic control while reducing circulating insulin levels.
Pharmacological studies in animal models indicate that rosiglitazone improves sensitivity to insulin in muscle and adipose tissue and inhibits hepatic gluconeogenesis. Rosiglitazone maleate is not chemically or functionally related to the sulfonylureas, the biguanides, or the alpha-glucosidase inhibitors.
Chemically, rosiglitazone maleate is (±)-5-[[4-[2-(methyl-2-pyridinylamino)ethoxy]phenyl]methyl]-2,4-thiazolidinedione, ( Z )-2-butenedioate (1:1) with a molecular weight of 473.52 (357.44 free base). The molecule has a single chiral center and is present as a racemate. Due to rapid interconversion, the enantiomers are functionally indistinguishable.
The molecular formula is C 18 H 19 N 3 O 3 S·C 4 H 4 O 4 . Rosiglitazone maleate is a white to off-white solid with a melting point range of 122° to 123°C. The pKa values of rosiglitazone maleate are 6.8 and 6.1. It is readily soluble in ethanol and a buffered aqueous solution with pH of 2.3; solubility decreases with increasing pH in the physiological range.
Each pentagonal film-coated TILTAB® tablet contains rosiglitazone maleate equivalent to rosiglitazone, 2 mg, 4 mg, or 8 mg, for oral administration. Inactive ingredients are: Hypromellose 2910, lactose monohydrate, magnesium stearate, microcrystalline cellulose, polyethylene glycol 3000, sodium starch glycolate, titanium dioxide, triacetin, and 1 or more of the following: Synthetic red and yellow iron oxides and talc.
CLINICAL PHARMACOLOGY
Mechanism of Action: Rosiglitazone, a member of the thiazolidinedione class of antidiabetic agents, improves glycemic control by improving insulin sensitivity. Rosiglitazone is a highly selective and potent agonist for the peroxisome proliferator-activated receptor-gamma (PPAR(gamma)). In humans, PPAR receptors are found in key target tissues for insulin action such as adipose tissue, skeletal muscle, and liver. Activation of PPAR(gamma) nuclear receptors regulates the transcription of insulin-responsive genes involved in the control of glucose production, transport, and utilization. In addition, PPAR(gamma)-responsive genes also participate in the regulation of fatty acid metabolism.
Insulin resistance is a common feature characterizing the pathogenesis of type 2 diabetes. The antidiabetic activity of rosiglitazone has been demonstrated in animal models of type 2 diabetes in which hyperglycemia and/or impaired glucose tolerance is a consequence of insulin resistance in target tissues. Rosiglitazone reduces blood glucose concentrations and reduces hyperinsulinemia in the ob/ob obese mouse, db/db diabetic mouse, and fa/fa fatty Zucker rat.
In animal models, rosiglitazone's antidiabetic activity was shown to be mediated by increased sensitivity to insulin's action in the liver, muscle, and adipose tissues. The expression of the insulin-regulated glucose transporter GLUT-4 was increased in adipose tissue. Rosiglitazone did not induce hypoglycemia in animal models of type 2 diabetes and/or impaired glucose tolerance.
Pharmacokinetics and Drug Metabolism: Maximum plasma concentration (C max ) and the area under the curve (AUC) of rosiglitazone increase in a dose-proportional manner over the therapeutic dose range (see Table 1). The elimination half-life is 3 to 4 hours and is independent of dose.
Table 1. Mean (SD) Pharmacokinetic Parameters for Rosiglitazone Following Single Oral Doses (N = 32) Parameter 1 mg
Fasting2 mg
Fasting8 mg
Fasting8 mg
FedAUC 0-inf
[ng.hr./mL]358
(112)733
(184)2,971
(730)2,890
(795)C max
[ng/mL]76
(13)156
(42)598
(117)432
(92)Half-life
[hr.]3.16
(0.72)3.15
(0.39)3.37
(0.63)3.59
(0.70)CL/F *
[L/hr.]3.03
(0.87)2.89
(0.71)2.85
(0.69)2.97
(0.81)*CL/F = Oral clearance. Absorption: The absolute bioavailability of rosiglitazone is 99%. Peak plasma concentrations are observed about 1 hour after dosing. Administration of rosiglitazone with food resulted in no change in overall exposure (AUC), but there was an approximately 28% decrease in C max and a delay in T max (1.75 hours). These changes are not likely to be clinically significant; therefore, AVANDIA may be administered with or without food.
Distribution: The mean (CV%) oral volume of distribution (Vss/F) of rosiglitazone is approximately 17.6 (30%) liters, based on a population pharmacokinetic analysis. Rosiglitazone is approximately 99.8% bound to plasma proteins, primarily albumin.
Metabolism: Rosiglitazone is extensively metabolized with no unchanged drug excreted in the urine. The major routes of metabolism were N-demethylation and hydroxylation, followed by conjugation with sulfate and glucuronic acid. All the circulating metabolites are considerably less potent than parent and, therefore, are not expected to contribute to the insulin-sensitizing activity of rosiglitazone.
In vitro data demonstrate that rosiglitazone is predominantly metabolized by Cytochrome P 450 (CYP) isoenzyme 2C8, with CYP2C9 contributing as a minor pathway.
Excretion: Following oral or intravenous administration of [ 14 C]rosiglitazone maleate, approximately 64% and 23% of the dose was eliminated in the urine and in the feces, respectively. The plasma half-life of [ 14 C]related material ranged from 103 to 158 hours.
Population Pharmacokinetics in Patients with Type 2 Diabetes: Population pharmacokinetic analyses from 3 large clinical trials including 642 men and 405 women with type 2 diabetes (aged 35 to 80 years) showed that the pharmacokinetics of rosiglitazone are not influenced by age, race, smoking, or alcohol consumption. Both oral clearance (CL/F) and oral steady-state volume of distribution (Vss/F) were shown to increase with increases in body weight. Over the weight range observed in these analyses (50 to 150 kg), the range of predicted CL/F and Vss/F values varied by <1.7-fold and <2.3-fold, respectively. Additionally, rosiglitazone CL/F was shown to be influenced by both weight and gender, being lower (about 15%) in female patients.
Special Populations: Geriatric: Results of the population pharmacokinetic analysis (n = 716 <65 years; n = 331 >/=65 years) showed that age does not significantly affect the pharmacokinetics of rosiglitazone.
Gender: Results of the population pharmacokinetics analysis showed that the mean oral clearance of rosiglitazone in female patients (n = 405) was approximately 6% lower compared to male patients of the same body weight (n = 642).
As monotherapy and in combination with metformin, AVANDIA improved glycemic control in both males and females. In metformin combination studies, efficacy was demonstrated with no gender differences in glycemic response.
In monotherapy studies, a greater therapeutic response was observed in females; however, in more obese patients, gender differences were less evident. For a given body mass index (BMI), females tend to have a greater fat mass than males. Since the molecular target PPAR(gamma) is expressed in adipose tissues, this differentiating characteristic may account, at least in part, for the greater response to AVANDIA in females. Since therapy should be individualized, no dose adjustments are necessary based on gender alone.
Hepatic Impairment: Unbound oral clearance of rosiglitazone was significantly lower in patients with moderate to severe liver disease (Child-Pugh Class B/C) compared to healthy subjects. As a result, unbound C max and AUC 0-inf were increased 2- and 3-fold, respectively. Elimination half-life for rosiglitazone was about 2 hours longer in patients with liver disease, compared to healthy subjects.
Therapy with AVANDIA should not be initiated if the patient exhibits clinical evidence of active liver disease or increased serum transaminase levels (ALT >2.5 × upper limit of normal) at baseline (see PRECAUTIONS , General , Hepatic Effects ).
Renal Impairment: There are no clinically relevant differences in the pharmacokinetics of rosiglitazone in patients with mild to severe renal impairment or in hemodialysis-dependent patients compared to subjects with normal renal function. No dosage adjustment is therefore required in such patients receiving AVANDIA. Since metformin is contraindicated in patients with renal impairment, coadministration of metformin with AVANDIA is contraindicated in these patients.
Race: Results of a population pharmacokinetic analysis including subjects of Caucasian, black, and other ethnic origins indicate that race has no influence on the pharmacokinetics of rosiglitazone.
Drug Interactions:
Drugs that Inhibit, Induce, or are Metabolized by Cytochrome P 450 : In vitro drug metabolism studies suggest that rosiglitazone does not inhibit any of the major P 450 enzymes at clinically relevant concentrations. In vitro data demonstrate that rosiglitazone is predominantly metabolized by CYP2C8, and to a lesser extent, 2C9.
Gemfibrozil: Concomitant administration of gemfibrozil (600 mg twice daily), an inhibitor of CYP2C8, and rosiglitazone (4 mg once daily) for 7 days increased rosiglitazone AUC by 127%, compared to the administration of rosiglitazone (4 mg once daily) alone. Given the potential for dose-related adverse events with rosiglitazone, a decrease in the dose of rosiglitazone may be needed when gemfibrozil is introduced (see PRECAUTIONS ).
Rifampin: Rifampin administration (600 mg once a day), an inducer of CYP2C8, for 6 days is reported to decrease rosiglitazone AUC by 66%, compared to the administration of rosiglitazone (8 mg) alone (see PRECAUTIONS ). 1
AVANDIA (4 mg twice daily) was shown to have no clinically relevant effect on the pharmacokinetics of nifedipine and oral contraceptives (ethinyl estradiol and norethindrone), which are predominantly metabolized by CYP3A4.
Glyburide: AVANDIA (2 mg twice daily) taken concomitantly with glyburide (3.75 to 10 mg/day) for 7 days did not alter the mean steady-state 24-hour plasma glucose concentrations in diabetic patients stabilized on glyburide therapy. Repeat doses of AVANDIA (8 mg once daily) for 8 days in healthy adult Caucasian subjects caused a decrease in glyburide AUC (32%) and C max (35%). In Japanese subjects, glyburide AUC (14%) and C max (31%) slightly increased following coadministration of AVANDIA.
Glimepiride: Single oral doses of glimepiride in 14 healthy adult subjects had no clinically significant effect on the steady-state pharmacokinetics of AVANDIA. No clinically significant reductions in glimepiride AUC and C max were observed after repeat doses of AVANDIA (8 mg once daily) for 8 days in healthy adult subjects.
Metformin: Concurrent administration of AVANDIA (2 mg twice daily) and metformin (500 mg twice daily) in healthy volunteers for 4 days had no effect on the steady-state pharmacokinetics of either metformin or rosiglitazone.
Acarbose: Coadministration of acarbose (100 mg three times daily) for 7 days in healthy volunteers had no clinically relevant effect on the pharmacokinetics of a single oral dose of AVANDIA.
Digoxin: Repeat oral dosing of AVANDIA (8 mg once daily) for 14 days did not alter the steady-state pharmacokinetics of digoxin (0.375 mg once daily) in healthy volunteers.
Warfarin: Repeat dosing with AVANDIA had no clinically relevant effect on the steady-state pharmacokinetics of warfarin enantiomers.
Ethanol: A single administration of a moderate amount of alcohol did not increase the risk of acute hypoglycemia in type 2 diabetes mellitus patients treated with AVANDIA.
Ranitidine: Pretreatment with ranitidine (150 mg twice daily for 4 days) did not alter the pharmacokinetics of either single oral or intravenous doses of rosiglitazone in healthy volunteers. These results suggest that the absorption of oral rosiglitazone is not altered in conditions accompanied by increases in gastrointestinal pH.
CLINICAL STUDIES
In clinical studies, treatment with AVANDIA resulted in an improvement in glycemic control, as measured by fasting plasma glucose (FPG) and hemoglobin A1c (HbA1c), with a concurrent reduction in insulin and C-peptide. Postprandial glucose and insulin were also reduced. This is consistent with the mechanism of action of AVANDIA as an insulin sensitizer. The improvement in glycemic control was durable, with maintenance of effect for 52 weeks. The maximum recommended daily dose is 8 mg. Dose-ranging studies suggested that no additional benefit was obtained with a total daily dose of 12 mg.
The addition of AVANDIA to either metformin, a sulfonylurea, or insulin resulted in significant reductions in hyperglycemia compared to any of these agents alone. These results are consistent with an additive effect on glycemic control when AVANDIA is used as combination therapy.
Patients with lipid abnormalities were not excluded from clinical trials of AVANDIA. In all 26-week controlled trials, across the recommended dose range, AVANDIA as monotherapy was associated with increases in total cholesterol, LDL, and HDL and decreases in free fatty acids. These changes were statistically significantly different from placebo or glyburide controls (see Table 2).
Increases in LDL occurred primarily during the first 1 to 2 months of therapy with AVANDIA and LDL levels remained elevated above baseline throughout the trials. In contrast, HDL continued to rise over time. As a result, the LDL/HDL ratio peaked after 2 months of therapy and then appeared to decrease over time. Because of the temporal nature of lipid changes, the 52-week glyburide-controlled study is most pertinent to assess long-term effects on lipids. At baseline, week 26, and week 52, mean LDL/HDL ratios were 3.1, 3.2, and 3.0, respectively, for AVANDIA 4 mg twice daily. The corresponding values for glyburide were 3.2, 3.1, and 2.9. The differences in change from baseline between AVANDIA and glyburide at week 52 were statistically significant.
The pattern of LDL and HDL changes following therapy with AVANDIA in combination with other hypoglycemic agents were generally similar to those seen with AVANDIA in monotherapy.
The changes in triglycerides during therapy with AVANDIA were variable and were generally not statistically different from placebo or glyburide controls.
Table 2. Summary of Mean Lipid Changes in 26-Week Placebo-Controlled and 52-Week Glyburide-Controlled Monotherapy Studies Placebo-controlled Studies
Week 26Glyburide-controlled Study
Week 26 and Week 52Placebo AVANDIA Glyburide Titration AVANDIA 8 mg 4 mg
daily *8 mg
daily *Wk 26 Wk 52 Wk 26 Wk 52 Free Fatty Acids
N207
428
436
181
168 166 145 Baseline (mean) 18.1 17.5 17.9- 26.4 26.4 26.9 26.6 % Change from
baseline (mean)+0.2% -7.8% 14.7% -2.4% -4.7% -20.8% -21.5% LDL
N190
400
374
175
160 161 133 Baseline (mean) 123.7 126.8 125.3 142.7 141.9 142.1 142.1 % Change from
baseline (mean)+4.8% +14.1% +18.6% -0.9% -0.5% +11.9% +12.1% HDL
N208
429
436
184
170 170 145 Baseline (mean) 44.1 44.4 43.0 47.2 47.7 48.4 48.3 % Change from
baseline (mean)+8.0% +11.4% +14.2% +4.3% +8.7% +14.0% +18.5% *Once daily and twice daily dosing groups were combined. Monotherapy: A total of 2,315 patients with type 2 diabetes, previously treated with diet alone or antidiabetic medication(s), were treated with AVANDIA as monotherapy in 6 double-blind studies, which included two 26-week placebo-controlled studies, one 52-week glyburide-controlled study, and 3 placebo-controlled dose-ranging studies of 8 to 12 weeks duration. Previous antidiabetic medication(s) were withdrawn and patients entered a 2 to 4 week placebo run-in period prior to randomization.
Two 26-week, double-blind, placebo-controlled trials, in patients with type 2 diabetes (n = 1,401) with inadequate glycemic control (mean baseline FPG approximately 228 mg/dL [101 to 425 mg/dL] and mean baseline HbA1c 8.9% [5.2% to 16.2%]), were conducted. Treatment with AVANDIA produced statistically significant improvements in FPG and HbA1c compared to baseline and relative to placebo. Data from one of these studies are summarized in Table 3.
Table 3. Glycemic Parameters in a 26-Week Placebo-Controlled Trial Placebo AVANDIA AVANDIA 4 mg once
daily2 mg twice
daily8 mg once
daily4 mg twice
dailyN 173 180 186 181 187 FPG (mg/dL)
Baseline (mean)225 229 225 228 228 Change from baseline (mean) 8 -25 -35 -42 -55 Difference from placebo
(adjusted mean)- -31 * -43 * -49 * -62 * % of patients with >/=30 mg/dL
decrease from baseline19% 45% 54% 58% 70% HbA1c (%)
Baseline (mean)8.9 8.9 8.9 8.9 9.0 Change from baseline (mean) 0.8 0.0 -0.1 -0.3 -0.7 Difference from placebo
(adjusted mean)- -0.8 * -0.9 * -1.1 * -1.5 * % of patients with >/=0.7%
decrease from baseline9% 28% 29% 39% 54% *<0.0001 compared to placebo. When administered at the same total daily dose, AVANDIA was generally more effective in reducing FPG and HbA1c when administered in divided doses twice daily compared to once daily doses. However, for HbA1c, the difference between the 4 mg once daily and 2 mg twice daily doses was not statistically significant.
Long-term maintenance of effect was evaluated in a 52-week, double-blind, glyburide-controlled trial in patients with type 2 diabetes. Patients were randomized to treatment with AVANDIA 2 mg twice daily (N = 195) or AVANDIA 4 mg twice daily (N = 189) or glyburide (N = 202) for 52 weeks. Patients receiving glyburide were given an initial dosage of either 2.5 mg/day or 5.0 mg/day. The dosage was then titrated in 2.5 mg/day increments over the next 12 weeks, to a maximum dosage of 15.0 mg/day in order to optimize glycemic control. Thereafter the glyburide dose was kept constant.
The median titrated dose of glyburide was 7.5 mg. All treatments resulted in a statistically significant improvement in glycemic control from baseline (see Figure 1 and Figure 2). At the end of week 52, the reduction from baseline in FPG and HbA1c was -40.8 mg/dL and -0.53% with AVANDIA 4 mg twice daily; -25.4 mg/dL and -0.27% with AVANDIA 2 mg twice daily; and -30.0 mg/dL and -0.72% with glyburide. For HbA1c, the difference between AVANDIA 4 mg twice daily and glyburide was not statistically significant at week 52. The initial fall in FPG with glyburide was greater than with AVANDIA; however, this effect was less durable over time. The improvement in glycemic control seen with AVANDIA 4 mg twice daily at week 26 was maintained through week 52 of the study.
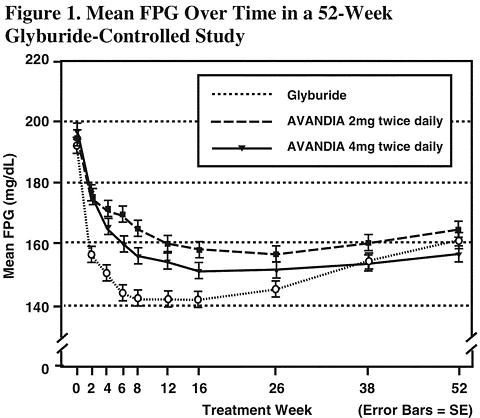
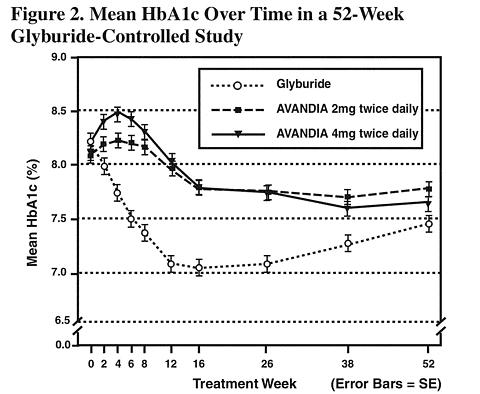
Hypoglycemia was reported in 12.1% of glyburide-treated patients versus 0.5% (2 mg twice daily) and 1.6% (4 mg twice daily) of patients treated with AVANDIA. The improvements in glycemic control were associated with a mean weight gain of 1.75 kg and 2.95 kg for patients treated with 2 mg and 4 mg twice daily of AVANDIA, respectively, versus 1.9 kg in glyburide-treated patients. In patients treated with AVANDIA, C-peptide, insulin, pro-insulin, and pro-insulin split products were significantly reduced in a dose-ordered fashion, compared to an increase in the glyburide-treated patients.
Combination With Metformin: A total of 670 patients with type 2 diabetes participated in two 26-week, randomized, double-blind, placebo/active-controlled studies designed to assess the efficacy of AVANDIA in combination with metformin. AVANDIA, administered in either once daily or twice daily dosing regimens, was added to the therapy of patients who were inadequately controlled on a maximum dose (2.5 grams/day) of metformin.
In one study, patients inadequately controlled on 2.5 grams/day of metformin (mean baseline FPG 216 mg/dL and mean baseline HbA1c 8.8%) were randomized to receive 4 mg of AVANDIA once daily, 8 mg of AVANDIA once daily, or placebo in addition to metformin. A statistically significant improvement in FPG and HbA1c was observed in patients treated with the combinations of metformin and 4 mg of AVANDIA once daily and 8 mg of AVANDIA once daily, versus patients continued on metformin alone (see Table 4).
Table 4. Glycemic Parameters in a 26-Week
Combination Study of AVANDIA Plus MetforminMetformin AVANDIA
4 mg once daily
+ metforminAVANDIA
8 mg once daily
+ metforminN 113 116 110 FPG (mg/dL)
Baseline (mean)214 215 220 Change from baseline (mean) 6 -33 -48 Difference from metformin alone
(adjusted mean)- -40 * -53 * % of patients with >/=30 mg/dL
decrease from baseline20% 45% 61% HbA1c (%)
Baseline (mean)8.6 8.9 8.9 Change from baseline (mean) 0.5 -0.6 -0.8 Difference from metformin alone
(adjusted mean)- -1.0 * -1.2 * % of patients with >/=0.7%
decrease from baseline11% 45% 52% *<0.0001 compared to metformin. In a second 26-week study, patients with type 2 diabetes inadequately controlled on 2.5 grams/day of metformin who were randomized to receive the combination of AVANDIA 4 mg twice daily and metformin (N = 105) showed a statistically significant improvement in glycemic control with a mean treatment effect for FPG of -56 mg/dL and a mean treatment effect for HbA1c of -0.8% over metformin alone. The combination of metformin and AVANDIA resulted in lower levels of FPG and HbA1c than either agent alone.
Patients who were inadequately controlled on a maximum dose (2.5 grams/day) of metformin and who were switched to monotherapy with AVANDIA demonstrated loss of glycemic control, as evidenced by increases in FPG and HbA1c. In this group, increases in LDL and VLDL were also seen.
Combination With a Sulfonylurea: A total of 3,457 patients with type 2 diabetes participated in ten 24- to 26-week randomized, double-blind, placebo/active-controlled studies and one 2-year double-blind, active-controlled study in elderly patients designed to assess the efficacy and safety of AVANDIA in combination with a sulfonylurea. AVANDIA 2 mg, 4 mg, or 8 mg daily, was administered either once daily (3 studies) or in divided doses twice daily (7 studies), to patients inadequately controlled on a submaximal or maximal dose of sulfonylurea.
In these studies, the combination of AVANDIA 4 mg or 8 mg daily (administered as single or twice daily divided doses) and a sulfonylurea significantly reduced FPG and HbA1c compared to placebo plus sulfonylurea or further up-titration of the sulfonylurea. Table 5 shows pooled data for 8 studies in which AVANDIA added to sulfonylurea was compared to placebo plus sulfonylurea.
Table 5. Glycemic Parameters in 24- to 26-Week Combination Studies of AVANDIA Plus Sulfonylurea Twice Daily Divided Dosing
(5 Studies)Sulfonylurea AVANDIA
2 mg twice
daily +
sulfonylureaSulfonylurea AVANDIA
4 mg twice
daily +
sulfonylureaN 397 497 248 346 FPG (mg/dL) Baseline (mean) 204 198 188 187 Change from baseline (mean) 11 -29 8 -43 Difference from sulfonylurea
alone (adjusted mean)- -42 * - -53 * % of patients with >/=30 mg/dL
decrease from baseline17% 49% 15% 61% HbA1c (%) Baseline (mean) 9.4 9.5 9.3 9.6 Change from baseline (mean) 0.2 -1.0 0.0 -1.6 Difference from sulfonylurea
alone (adjusted mean)- -1.1 * - -1.4 * % of patients with >/=0.7%
decrease from baseline21% 60% 23% 75% Once Daily Dosing
(3 Studies)Sulfonylurea AVANDIA
4 mg once
daily +
sulfonylureaSulfonylurea AVANDIA
8 mg once
daily +
sulfonylureaN 172 172 173 176 FPG (mg/dL) Baseline (mean) 198 206 188 192 Change from baseline (mean) 17 -25 17 -43 Difference from sulfonylurea
alone (adjusted mean)- -47 * - -66 * % of patients with >/=30 mg/dL
decrease from baseline17% 48% 19% 55% HbA1c (%) Baseline (mean) 8.6 8.8 8.9 8.9 Change from baseline (mean) 0.4 -0.5 0.1 -1.2 Difference from sulfonylurea
alone (adjusted mean)- -0.9 * - -1.4 * % of patients with >/=0.7%
decrease from baseline11% 36% 20% 68% *<0.0001 compared to sulfonylurea alone. One of the 24- to 26-week studies included patients who were inadequately controlled on maximal doses of glyburide and switched to 4 mg of AVANDIA daily as monotherapy; in this group, loss of glycemic control was demonstrated, as evidenced by increases in FPG and HbA1c.
In a 2-year double-blind study, elderly patients (aged 59 to 89 years) on half-maximal sulfonylurea (glipizide 10 mg twice daily) were randomized to the addition of AVANDIA (n = 115, 4 mg once daily to 8 mg as needed) or to continued up-titration of glipizide (n = 110), to a maximum of 20 mg twice daily. Mean baseline FPG and HbA1c were 157 mg/dL and 7.72%, respectively, for the AVANDIA plus glipizide arm and 159 mg/dL and 7.65%, respectively, for the glipizide uptitration arm. Loss of glycemic control (FPG >/=180 mg/dL) occurred in a significantly lower proportion of patients (2%) on AVANDIA plus glipizide compared to patients in the glipizide up-titration arm (28.7%). About 78% of the patients on combination therapy completed the 2 years of therapy while only 51% completed on glipizide monotherapy. The effect of combination therapy on FPG and HbA1c was durable over the 2-year study period, with patients achieving a mean of 132 mg/dL for FPG and a mean of 6.98% for HbA1c compared to no change on the glipizide arm.
Combination With Insulin: In two 26-week randomized, double-blind, fixed-dose studies designed to assess the efficacy and safety of AVANDIA in combination with insulin, patients inadequately controlled on insulin (65 to 76 units/day, mean range at baseline) were randomized to receive AVANDIA 4 mg plus insulin (n = 206) or placebo plus insulin (n = 203). The mean duration of disease in these patients was 12 to 13 years.
Compared to insulin plus placebo, single or divided doses of AVANDIA 4 mg daily plus insulin significantly reduced FPG (mean reduction of 32 to 40 mg/dL) and HbA1c (mean reduction of 0.6% to 0.7%). Approximately 40% of all patients treated with AVANDIA reduced their insulin dose.
Combination With Sulfonylurea and Metformin: In two 24- to 26-week, double-blind, placebo-controlled, studies designed to assess the efficacy and safety of AVANDIA in combination with sulfonylurea plus metformin, AVANDIA 4 mg or 8 mg daily, was administered in divided doses twice daily, to patients inadequately controlled on submaximal (10 mg) and maximal (20 mg) doses of glyburide and maximal dose of metformin (2 g/day). A statistically significant improvement in FPG and HbA1c was observed in patients treated with the combinations of sulfonylurea plus metformin and 4 mg of AVANDIA and 8 mg of AVANDIA versus patients continued on sulfonylurea plus metformin, as shown in Table 6.
Table 6. Glycemic Parameters in a 26-Week Combination
Study of AVANDIA Plus Sulfonylurea and MetforminSulfonylurea +
metforminAVANDIA
2 mg twice daily +
sulfonylurea +
metforminAVANDIA
4 mg twice daily +
sulfonylurea +
metforminN 273 276 277 FPG (mg/dL) Baseline (mean) 189 190 192 Change from baseline (mean) 14 -19 -40 Difference from sulfonylurea
plus metformin (adjusted mean)- -30 * -52 * % of patients with >/=30 mg/dL
decrease from baseline16% 46% 62% HbA1c (%) Baseline (mean) 8.7 8.6 8.7 Change from baseline (mean) 0.2 -0.4 -0.9 Difference from sulfonylurea
plus metformin (adjusted mean)- -0.6 * -1.1 * % of patients with >/=0.7%
decrease from baseline16% 39% 63% *<0.0001 compared to placebo. INDICATIONS AND USAGE
AVANDIA is indicated as an adjunct to diet and exercise to improve glycemic control in patients with type 2 diabetes mellitus.
- AVANDIA is indicated as monotherapy.
- AVANDIA is also indicated for use in combination with a sulfonylurea, metformin, or insulin when diet, exercise, and a single agent do not result in adequate glycemic control. For patients inadequately controlled with a maximum dose of a sulfonylurea or metformin, AVANDIA should be added to, rather than substituted for, a sulfonylurea or metformin.
- AVANDIA is also indicated for use in combination with a sulfonylurea plus metformin when diet, exercise, and both agents do not result in adequate glycemic control.
Management of type 2 diabetes should include diet control. Caloric restriction, weight loss, and exercise are essential for the proper treatment of the diabetic patient because they help improve insulin sensitivity. This is important not only in the primary treatment of type 2 diabetes, but also in maintaining the efficacy of drug therapy. Prior to initiation of therapy with AVANDIA, secondary causes of poor glycemic control, e.g., infection, should be investigated and treated.
CONTRAINDICATIONS
AVANDIA is contraindicated in patients with known hypersensitivity to this product or any of its components.
WARNINGS
Cardiac Failure and Other Cardiac Effects: AVANDIA, like other thiazolidinediones, alone or in combination with other antidiabetic agents, can cause fluid retention, which may exacerbate or lead to heart failure. Patients should be observed for signs and symptoms of heart failure. In combination with insulin, thiazolidinediones may also increase the risk of other cardiovascular adverse events. AVANDIA should be discontinued if any deterioration in cardiac status occurs.
Patients with New York Heart Association (NYHA) Class 3 and 4 cardiac status were not studied during the clinical trials. AVANDIA is not recommended in patients with NYHA Class 3 and 4 cardiac status.
In three 26-week trials in patients with type 2 diabetes, 216 received 4 mg of AVANDIA plus insulin, 322 received 8 mg of AVANDIA plus insulin, and 338 received insulin alone. These trials included patients with long-standing diabetes and a high prevalence of pre-existing medical conditions, including peripheral neuropathy, retinopathy, ischemic heart disease, vascular disease, and congestive heart failure. In these clinical studies an increased incidence of edema, cardiac failure, and other cardiovascular adverse events was seen in patients on AVANDIA and insulin combination therapy compared to insulin and placebo. Patients who experienced cardiovascular events were on average older and had a longer duration of diabetes. These cardiovascular events were noted at both the 4 mg and 8 mg daily doses of AVANDIA. In this population, however, it was not possible to determine specific risk factors that could be used to identify all patients at risk of heart failure and other cardiovascular events on combination therapy. Three of 10 patients who developed cardiac failure on combination therapy during the double-blind part of the fixed-dose studies had no known prior evidence of congestive heart failure, or pre-existing cardiac condition.
In a double-blind study in type 2 diabetes patients with chronic renal failure (112 received 4 mg or 8 mg of AVANDIA plus insulin and 108 received insulin control), there was no difference in cardiovascular adverse events with AVANDIA in combination with insulin compared to insulin control.
Patients treated with combination AVANDIA and insulin should be monitored for cardiovascular adverse events. This combination therapy should be discontinued in patients who do not respond as manifested by a reduction in HbA1c or insulin dose after 4 to 5 months of therapy or who de-velop any significant adverse events. (See ADVERSE REACTIONS ).
PRECAUTIONS
General: Due to its mechanism of action, AVANDIA is active only in the presence of endogenous insulin. Therefore, AVANDIA should not be used in patients with type 1 diabetes or for the treatment of diabetic ketoacidosis.
Hypoglycemia: Patients receiving AVANDIA in combination with other hypoglycemic agents may be at risk for hypoglycemia, and a reduction in the dose of the concomitant agent may be necessary.
Edema: AVANDIA should be used with caution in patients with edema. In a clinical study in healthy volunteers who received 8 mg of AVANDIA once daily for 8 weeks, there was a statistically significant increase in median plasma volume compared to placebo.
Since thiazolidinediones, including rosiglitazone, can cause fluid retention, which can exacerbate or lead to congestive heart failure, AVANDIA should be used with caution in patients at risk for heart failure. Patients should be monitored for signs and symptoms of heart failure (see WARNINGS , Cardiac Failure and Other Cardiac Effects and PRECAUTIONS , Information for Patients ).
In controlled clinical trials of patients with type 2 diabetes, mild to moderate edema was reported in patients treated with AVANDIA, and may be dose related. Patients with ongoing edema are more likely to have adverse events associated with edema if started on combination therapy with insulin and AVANDIA (see ADVERSE REACTIONS ).
Weight Gain: Dose-related weight gain was seen with AVANDIA alone and in combination with other hypoglycemic agents (see Table 7). The mechanism of weight gain is unclear but probably involves a combination of fluid retention and fat accumulation.
In postmarketing experience, there have been reports of unusually rapid increases in weight and increases in excess of that generally observed in clinical trials. Patients who experience such increases should be assessed for fluid accumulation and volume-related events such as excessive edema and congestive heart failure.
Table 7. Weight Changes (kg) From Baseline During Clinical Trials With AVANDIA Control Group AVANDIA
4 mgAVANDIA
8 mgMonotherapy Duration Median
(25 th , 75 th
percentile)Median
(25 th , 75 th
percentile)Median
(25 th , 75 th
percentile)26 weeks placebo -0.9 (-2.8, 0.9)
n = 2101.0 (-0.9, 3.6)
n = 4363.1 (1.1, 5.8)
n = 43952 weeks sulfonylurea 2.0 (0, 4.0)
n = 1732.0 (-0.6, 4.0)
n = 1502.6 (0, 5.3)
n = 157Combination therapy sulfonylurea 24-26 weeks sulfonylurea 0 (-1.0, 1.3)
n = 1,1552.2 (0.5, 4.0)
n = 6133.5 (1.4, 5.9)
n = 841metformin 26 weeks metformin -1.4 (-3.2, 0.2)
n = 1750.8 (-1.0, 2.6)
n = 1002.1 (0, 4.3)
n = 184insulin 26 weeks insulin 0.9 (-0.5, 2.7)
n = 1624.1 (1.4, 6.3)
n = 1645.4 (3.4, 7.3)
n = 150sulfonylurea + metformin 26 weeks sulfonylurea
+ metformin0.2 (-1.2, 1.6)
n = 2722.5 (0.8, 4.6)
n = 2754.5 (2.4, 7.3)
n = 276Hematologic: Across all controlled clinical studies, decreases in hemoglobin and hematocrit (mean decreases in individual studies </=1.0 gram/dL and </=3.3%, respectively) were observed for AVANDIA alone and in combination with other hypoglycemic agents. The changes occurred primarily during the first 3 months following initiation of therapy with AVANDIA or following a dose increase in AVANDIA. White blood cell counts also decreased slightly in patients treated with AVANDIA. The observed changes may be related to the increased plasma volume observed with treatment with AVANDIA and may be dose related (see ADVERSE REACTIONS , Laboratory Abnormalities, Hematologic ).
Ovulation: Therapy with AVANDIA, like other thiazolidinediones, may result in ovulation in some premenopausal anovulatory women. As a result, these patients may be at an increased risk for pregnancy while taking AVANDIA (see PRECAUTIONS , Pregnancy, Pregnancy Category C ). Thus, adequate contraception in premenopausal women should be recommended. This possible effect has not been specifically investigated in clinical studies so the frequency of this occurrence is not known.
Although hormonal imbalance has been seen in preclinical studies (see PRECAUTIONS , Carcinogenesis, Mutagenesis, Impairment of Fertility ), the clinical significance of this finding is not known. If unexpected menstrual dysfunction occurs, the benefits of continued therapy with AVANDIA should be reviewed.
Hepatic Effects: Another drug of the thiazolidinedione class, troglitazone, was associated with idiosyncratic hepatotoxicity, and very rare cases of liver failure, liver transplants, and death were reported during clinical use. In pre-approval controlled clinical trials in patients with type 2 diabetes, troglitazone was more frequently associated with clinically significant elevations in liver enzymes (ALT >3 × upper limit of normal) compared to placebo. Very rare cases of reversible jaundice were also reported.
In pre-approval clinical studies in 4,598 patients treated with AVANDIA, encompassing approximately 3,600 patient years of exposure, there was no signal of drug-induced hepatotoxicity or elevation of ALT levels. In the pre-approval controlled trials, 0.2% of patients treated with AVANDIA had elevations in ALT >3 × the upper limit of normal compared to 0.2% on placebo and 0.5% on active comparators. The ALT elevations in patients treated with AVANDIA were reversible and were not clearly causally related to therapy with AVANDIA.
In postmarketing experience with AVANDIA, reports of hepatitis and of hepatic enzyme elevations to 3 or more times the upper limit of normal have been received. Very rarely, these reports have involved hepatic failure with and without fatal outcome, although causality has not been established. Rosiglitazone is structurally related to troglitazone, a thiazolidinedione no longer marketed in the United States, which was associated with idiosyncratic hepatotoxicity and rare cases of liver failure, liver transplants, and death during clinical use. Pending the availability of the results of additional large, long-term controlled clinical trials and additional postmarketing safety data, it is recommended that patients treated with AVANDIA undergo periodic monitoring of liver enzymes.
Liver enzymes should be checked prior to the initiation of therapy with AVANDIA in all patients and periodically thereafter per the clinical judgement of the healthcare professional. Therapy with AVANDIA should not be initiated in patients with increased baseline liver enzyme levels (ALT >2.5 × upper limit of normal). Patients with mildly elevated liver enzymes (ALT levels </=2.5 × upper limit of normal) at baseline or during therapy with AVANDIA should be evaluated to determine the cause of the liver enzyme elevation. Initiation of, or continuation of, therapy with AVANDIA in patients with mild liver enzyme elevations should proceed with caution and include close clinical follow-up, including more frequent liver enzyme monitoring, to determine if the liver enzyme elevations resolve or worsen. If at any time ALT levels increase to >3 × the upper limit of normal in patients on therapy with AVANDIA, liver enzyme levels should be rechecked as soon as possible. If ALT levels remain >3 × the upper limit of normal, therapy with AVANDIA should be discontinued.
If any patient develops symptoms suggesting hepatic dysfunction, which may include unexplained nausea, vomiting, abdominal pain, fatigue, anorexia and/or dark urine, liver enzymes should be checked. The decision whether to continue the patient on therapy with AVANDIA should be guided by clinical judgment pending laboratory evalu-ations. If jaundice is observed, drug therapy should be discontinued.
There are no data available from clinical trials to evaluate the safety of AVANDIA in patients who experienced liver abnormalities, hepatic dysfunction, or jaundice while on troglitazone. AVANDIA should not be used in patients who experienced jaundice while taking troglitazone.
Laboratory Tests: Periodic fasting blood glucose and HbA1c measurements should be performed to monitor therapeutic response.
Liver enzyme monitoring is recommended prior to initiation of therapy with AVANDIA in all patients and periodically thereafter (see PRECAUTIONS , General , Hepatic Effects and ADVERSE REACTIONS , Laboratory Abnormalities , Serum Transaminase Levels ).
Information for Patients: Patients should be informed of the following: Management of type 2 diabetes should include diet control. Caloric restriction, weight loss, and exercise are essential for the proper treatment of the diabetic patient because they help improve insulin sensitivity. This is important not only in the primary treatment of type 2 diabetes, but in maintaining the efficacy of drug therapy.
It is important to adhere to dietary instructions and to regularly have blood glucose and glycosylated hemoglobin tested. Patients should be advised that it can take 2 weeks to see a reduction in blood glucose and 2 to 3 months to see full effect. Patients should be informed that blood will be drawn to check their liver function prior to the start of therapy and periodically thereafter per the clinical judgement of the healthcare professional. Patients with unexplained symptoms of nausea, vomiting, abdominal pain, fatigue, anorexia, or dark urine should immediately report these symptoms to their physician. Patients who experience an unusually rapid increase in weight or edema or who develop shortness of breath or other symptoms of heart failure while on AVANDIA should immediately report these symptoms to their physician.
AVANDIA can be taken with or without meals.
When using AVANDIA in combination with other hypoglycemic agents, the risk of hypoglycemia, its symptoms and treatment, and conditions that predispose to its development should be explained to patients and their family members.
Therapy with AVANDIA, like other thiazolidinediones, may result in ovulation in some premenopausal anovulatory women. As a result, these patients may be at an increased risk for pregnancy while taking AVANDIA (see PRECAUTIONS , Pregnancy, Pregnancy Category C ). Thus, adequate contraception in premenopausal women should be recommended. This possible effect has not been specifically investigated in clinical studies so the frequency of this occurrence is not known.
Drug Interactions: An inhibitor of CYP2C8 (such as gemfibrozil) may increase the AUC of rosiglitazone and an inducer of CYP2C8 (such as rifampin) may decrease the AUC of rosiglitazone. Therefore, if an inhibitor or an inducer of CYP2C8 is started or stopped during treatment with rosiglitazone, changes in diabetes treatment may be needed based upon clinical response. (See CLINICAL PHARMACOLOGY , Drug Interactions .)
Carcinogenesis, Mutagenesis, Impairment of Fertility: Carcinogenesis: A 2-year carcinogenicity study was conducted in Charles River CD-1 mice at doses of 0.4, 1.5, and 6 mg/kg/day in the diet (highest dose equivalent to approximately 12 times human AUC at the maximum recommended human daily dose). Sprague-Dawley rats were dosed for 2 years by oral gavage at doses of 0.05, 0.3, and 2 mg/kg/day (highest dose equivalent to approximately 10 and 20 times human AUC at the maximum recommended human daily dose for male and female rats, respectively).
Rosiglitazone was not carcinogenic in the mouse. There was an increase in incidence of adipose hyperplasia in the mouse at doses >/=1.5 mg/kg/day (approximately 2 times human AUC at the maximum recommended human daily dose). In rats, there was a significant increase in the incidence of benign adipose tissue tumors (lipomas) at doses >/=0.3 mg/kg/day (approximately 2 times human AUC at the maximum recommended human daily dose). These proliferative changes in both species are considered due to the persistent pharmacological overstimulation of adipose tissue.
Mutagenesis: Rosiglitazone was not mutagenic or clastogenic in the in vitro bacterial assays for gene mutation, the in vitro chromosome aberration test in human lymphocytes, the in vivo mouse micronucleus test, and the in vivo/in vitro rat UDS assay. There was a small (about 2-fold) increase in mutation in the in vitro mouse lymphoma assay in the presence of metabolic activation.
Impairment of Fertility: Rosiglitazone had no effects on mating or fertility of male rats given up to 40 mg/kg/day (approximately 116 times human AUC at the maximum recommended human daily dose). Rosiglitazone altered estrous cyclicity (2 mg/kg/day) and reduced fertility (40 mg/kg/day) of female rats in association with lower plasma levels of progesterone and estradiol (approximately 20 and 200 times human AUC at the maximum recommended human daily dose, respectively). No such effects were noted at 0.2 mg/kg/day (approximately 3 times human AUC at the maximum recommended human daily dose). In monkeys, rosiglitazone (0.6 and 4.6 mg/kg/day; approximately 3 and 15 times human AUC at the maximum recommended human daily dose, respectively) diminished the follicular phase rise in serum estradiol with consequential reduction in the luteinizing hormone surge, lower luteal phase progesterone levels, and amenorrhea. The mechanism for these effects appears to be direct inhibition of ovarian steroidogenesis.
Animal Toxicology: Heart weights were increased in mice (3 mg/kg/day), rats (5 mg/kg/day), and dogs (2 mg/kg/day) with rosiglitazone treatments (approximately 5, 22, and 2 times human AUC at the maximum recommended human daily dose, respectively). Morphometric measurement indicated that there was hypertrophy in cardiac ventricular tissues, which may be due to increased heart work as a result of plasma volume expansion.
Pregnancy: Pregnancy Category C: There was no effect on implantation or the embryo with rosiglitazone treatment during early pregnancy in rats, but treatment during mid-late gestation was associated with fetal death and growth retardation in both rats and rabbits. Teratogenicity was not observed at doses up to 3 mg/kg in rats and 100 mg/kg in rabbits (approximately 20 and 75 times human AUC at the maximum recommended human daily dose, respectively). Rosiglitazone caused placental pathology in rats (3 mg/kg/day). Treatment of rats during gestation through lactation reduced litter size, neonatal viability, and postnatal growth, with growth retardation reversible after puberty. For effects on the placenta, embryo/fetus, and offspring, the no-effect dose was 0.2 mg/kg/day in rats and 15 mg/kg/day in rabbits. These no-effect levels are approximately 4 times human AUC at the maximum recommended human daily dose.
There are no adequate and well-controlled studies in pregnant women. AVANDIA should not be used during pregnancy unless the potential benefit justifies the potential risk to the fetus.
Because current information strongly suggests that abnormal blood glucose levels during pregnancy are associated with a higher incidence of congenital anomalies as well as increased neonatal morbidity and mortality, most experts recommend that insulin monotherapy be used during pregnancy to maintain blood glucose levels as close to normal as possible.
Labor and Delivery: The effect of rosiglitazone on labor and delivery in humans is not known.
Nursing Mothers: Drug-related material was detected in milk from lactating rats. It is not known whether AVANDIA is excreted in human milk. Because many drugs are excreted in human milk, AVANDIA should not be administered to a nursing woman.
Pediatric Use: The safety and effectiveness of AVANDIA in pediatric patients have not been established.
Geriatric Use: Results of the population pharmacokinetic analysis showed that age does not significantly affect the pharmacokinetics of rosiglitazone (see CLINICAL PHARMACOLOGY , Special Populations ). Therefore, no dosage adjustments are required for the elderly. In controlled clinical trials, no overall differences in safety and effectiveness between older (>/=65 years) and younger (<65 years) patients were observed.
ADVERSE REACTIONS
In clinical trials, approximately 8,400 patients with type 2 diabetes have been treated with AVANDIA; 6,000 patients were treated for 6 months or longer and 3,000 patients were treated for 12 months or longer.
Trials of AVANDIA as Monotherapy and in Combination With Other Hypoglycemic Agents: The incidence and types of adverse events reported in clinical trials of AVANDIA as monotherapy are shown in Table 8.
Table 8. Adverse Events (>/=5% in Any Treatment Group) Reported by Patients in Double-blind Clinical Trials With
AVANDIA as MonotherapyPreferred Term AVANDIA
Monotherapy
N = 2,526Placebo
N = 601Metformin
N = 225Sulfonylureas *
N = 626% % % % Upper respiratory
tract infection9.9 8.7 8.9 7.3 Injury 7.6 4.3 7.6 6.1 Headache 5.9 5.0 8.9 5.4 Back pain 4.0 3.8 4.0 5.0 Hyperglycemia 3.9 5.7 4.4 8.1 Fatigue 3.6 5.0 4.0 1.9 Sinusitis 3.2 4.5 5.3 3.0 Diarrhea 2.3 3.3 15.6 3.0 Hypoglycemia 0.6 0.2 1.3 5.9 *Includes patients receiving glyburide (N = 514), gliclazide (N = 91) or glipizide (N = 21). Overall, the types of adverse experiences reported when AVANDIA was used in combination with a sulfonylurea or metformin were similar to those during monotherapy with AVANDIA. Events of anemia and edema tended to be reported more frequently at higher doses, and were generally mild to moderate in severity and usually did not require discontinuation of treatment with AVANDIA.
In double-blind studies, anemia was reported in 1.9% of patients receiving AVANDIA as monotherapy compared to 0.7% on placebo, 0.6% on sulfonylureas, and 2.2% on metformin. Reports of anemia were greater in patients treated with a combination of AVANDIA and metformin (7.1%) and with a combination of AVANDIA and a sulfonylurea plus metformin (6.7%) compared to monotherapy with AVANDIA or in combination with a sulfonylurea (2.3%). Lower pre-treatment hemoglobin/hematocrit levels in patients enrolled in the metformin combination clinical trials may have contributed to the higher reporting rate of anemia in these studies (see ADVERSE REACTIONS , Laboratory Abnormalities, Hematologic ).
In clinical trials, edema was reported in 4.8% of patients receiving AVANDIA as monotherapy compared to 1.3% on placebo, 1.0% on sulfonylureas, and 2.2% on metformin. The reporting rate of edema was higher for AVANDIA 8 mg in sulfonylurea combinations (12.4%) compared to other combinations, with the exception of insulin. Edema was reported in 14.7% of patients receiving AVANDIA in the insulin combination trials compared to 5.4% on insulin alone. Reports of new onset or exacerbation of congestive heart failure occurred at rates of 1% for insulin alone, and 2% (4 mg) and 3% (8 mg) for insulin in combination with AVANDIA.
In postmarketing experience in patients receiving thiazolidinedione therapy, serious adverse events with or without a fatal outcome, potentially related to volume expansion (e.g., congestive heart failure, pulmonary edema, and pleural effusions) have been reported. (See WARNINGS , Cardiac Failure and Other Cardiac Effects .)
In controlled combination therapy studies with sulfonylureas, mild to moderate hypoglycemic symptoms, which appear to be dose related, were reported. Few patients were withdrawn for hypoglycemia (<1%) and few episodes of hypoglycemia were considered to be severe (<1%). Hypoglycemia was the most frequently reported adverse event in the fixed-dose insulin combination trials, although few patients withdrew for hypoglycemia (4 of 408 for AVANDIA plus insulin and 1 of 203 for insulin alone). Rates of hypoglycemia, confirmed by capillary blood glucose concentration </=50 mg/dL, were 6% for insulin alone and 12% (4 mg) and 14% (8 mg) for insulin in combination with AVANDIA. (See PRECAUTIONS , General , Hypoglycemia and DOSAGE AND ADMINISTRATION , Combination Therapy .)
In postmarketing experience with AVANDIA, angioedema and urticaria have been reported rarely.
Laboratory Abnormalities: Hematologic: Decreases in mean hemoglobin and hematocrit occurred in a dose-related fashion in patients treated with AVANDIA (mean decreases in individual studies up to 1.0 gram/dL hemoglobin and up to 3.3% hematocrit). The time course and magnitude of decreases were similar in patients treated with a combination of AVANDIA and other hypoglycemic agents or AVANDIA monotherapy. Pre-treatment levels of hemoglobin and hematocrit were lower in patients in metformin combination studies and may have contributed to the higher reporting rate of anemia. White blood cell counts also decreased slightly in patients treated with AVANDIA. Decreases in hematologic parameters may be related to increased plasma volume observed with treatment with AVANDIA.
Lipids: Changes in serum lipids have been observed following treatment with AVANDIA (see CLINICAL STUDIES ).
Serum Transaminase Levels: In clinical studies in 4,598 patients treated with AVANDIA encompassing approximately 3,600 patient years of exposure, there was no evidence of drug-induced hepatotoxicity or elevated ALT levels.
In controlled trials, 0.2% of patients treated with AVANDIA had reversible elevations in ALT >3 × the upper limit of normal compared to 0.2% on placebo and 0.5% on active comparators. Hyperbilirubinemia was found in 0.3% of patients treated with AVANDIA compared with 0.9% treated with placebo and 1% in patients treated with active comparators.
In the clinical program including long-term, open-label experience, the rate per 100 patient years exposure of ALT increase to >3 × the upper limit of normal was 0.35 for patients treated with AVANDIA, 0.59 for placebo-treated patients, and 0.78 for patients treated with active comparator agents.
In pre-approval clinical trials, there were no cases of idiosyncratic drug reactions leading to hepatic failure. In postmarketing experience with AVANDIA, reports of hepatic enzyme elevations 3 or more times the upper limit of normal and hepatitis have been received (see PRECAUTIONS , General , Hepatic Effects ).
OVERDOSAGE
Limited data are available with regard to overdosage in humans. In clinical studies in volunteers, AVANDIA has been administered at single oral doses of up to 20 mg and was well-tolerated. In the event of an overdose, appropriate supportive treatment should be initiated as dictated by the patient's clinical status.
DOSAGE AND ADMINISTRATION
The management of antidiabetic therapy should be individualized. AVANDIA may be administered either at a starting dose of 4 mg as a single daily dose or divided and administered in the morning and evening. For patients who respond inadequately following 8 to 12 weeks of treatment, as determined by reduction in FPG, the dose may be increased to 8 mg daily as monotherapy or in combination with metformin, sulfonylurea, or sulfonylurea plus metformin. Reductions in glycemic parameters by dose and regimen are described under CLINICAL STUDIES . AVANDIA may be taken with or without food.
Monotherapy: The usual starting dose of AVANDIA is 4 mg administered either as a single dose once daily or in divided doses twice daily. In clinical trials, the 4 mg twice daily regimen resulted in the greatest reduction in FPG and HbA1c.
Combination Therapy: When AVANDIA is added to existing therapy, the current dose(s) of the agent(s) can be continued upon initiation of AVANDIA therapy.
Sulfonylurea: When used in combination with sulfonylurea, the usual starting dose of AVANDIA is 4 mg administered as either a single dose once daily or in divided doses twice daily. If patients report hypoglycemia, the dose of the sulfonylurea should be decreased.
Metformin: The usual starting dose of AVANDIA in combination with metformin is 4 mg administered as either a single dose once daily or in divided doses twice daily. It is unlikely that the dose of metformin will require adjustment due to hypoglycemia during combination therapy with AVANDIA.
Insulin: For patients stabilized on insulin, the insulin dose should be continued upon initiation of therapy with AVANDIA. AVANDIA should be dosed at 4 mg daily. Doses of AVANDIA greater than 4 mg daily in combination with insulin are not currently indicated. It is recommended that the insulin dose be decreased by 10% to 25% if the patient reports hypoglycemia or if FPG concentrations decrease to less than 100 mg/dL. Further adjustments should be individualized based on glucose-lowering response.
Sulfonylurea Plus Metformin: The usual starting dose of AVANDIA in combination with a sulfonylurea plus metformin is 4 mg administered as either a single dose once daily or divided doses twice daily. If patients report hypoglycemia, the dose of the sulfonylurea should be decreased.
Maximum Recommended Dose: The dose of AVANDIA should not exceed 8 mg daily, as a single dose or divided twice daily. The 8 mg daily dose has been shown to be safe and effective in clinical studies as monotherapy and in combination with metformin, sulfonylurea, or sulfonylurea plus metformin. Doses of AVANDIA greater than 4 mg daily in combination with insulin are not currently indicated.
AVANDIA may be taken with or without food.
No dosage adjustments are required for the elderly.
No dosage adjustment is necessary when AVANDIA is used as monotherapy in patients with renal impairment. Since metformin is contraindicated in such patients, concomitant administration of metformin and AVANDIA is also contraindicated in patients with renal impairment.
Therapy with AVANDIA should not be initiated if the patient exhibits clinical evidence of active liver disease or increased serum transaminase levels (ALT >2.5 × upper limit of normal at start of therapy) (see PRECAUTIONS , General , Hepatic Effects and CLINICAL PHARMACOLOGY , Special Populations , Hepatic Impairment ). Liver enzyme monitoring is recommended in all patients prior to initiation of therapy with AVANDIA and periodically thereafter (see PRECAUTIONS , General , Hepatic Effects ).
There are no data on the use of AVANDIA in patients younger than 18 years; therefore, use of AVANDIA in pediatric patients is not recommended.
HOW SUPPLIED
Tablets: Each pentagonal film-coated TILTAB tablet contains rosiglitazone as the maleate as follows: 2 mg-pink, debossed with SB on one side and 2 on the other; 4 mg-orange, debossed with SB on one side and 4 on the other; 8 mg-red-brown, debossed with SB on one side and 8 on the other.
2 mg bottles of 30: NDC 0029-3158-13
2 mg bottles of 60: NDC 0029-3158-18
2 mg bottles of 100: NDC 0029-3158-20
2 mg bottles of 500: NDC 0029-3158-25
2 mg SUP 100s: NDC 0029-3158-21
4 mg bottles of 30: NDC 0029-3159-13
4 mg bottles of 60: NDC 0029-3159-18
4 mg bottles of 100: NDC 0029-3159-20
4 mg bottles of 500: NDC 0029-3159-25
4 mg SUP 100s: NDC 0029-3159-21
8 mg bottles of 30: NDC 0029-3160-13
8 mg bottles of 100: NDC 0029-3160-20
8 mg bottles of 500: NDC 0029-3160-25
8 mg SUP 100s: NDC 0029-3160-21
STORAGE
Store at 25°C (77°F); excursions 15°-30°C (59°-86°F). Dispense in a tight, light-resistant container.
REFERENCE
- Park JY, Kim KA, Kang MH, et al. Effect of rifampin on the pharmacokinetics of rosiglitazone in healthy subjects. Clin Pharmacol Ther 2004;75:157-162.
Glaxosmithkline, Research Triangle Park, NC 27709
AVANDIA and TILTAB are registered trademarks of Glaxosmithkline.
©2005, Glaxosmithkline. All rights reserved.
March 2005/AV:L13
Subscribe to the "News" RSS Feed 
TOP ۞