-
Caduet Tablets (Pfizer)
DESCRIPTION
CADUET ® (amlodipine besylate and atorvastatin calcium) tablets combine the long-acting calcium channel blocker amlodipine besylate with the synthetic lipid-lowering agent atorvastatin calcium.
The amlodipine besylate component of CADUET is chemically described as 3-Ethyl-5-methyl (±)-2-[(2-amino-ethoxy)methyl]-4-(o-chlorophenyl)-1,4-dihydro-6-methyl-3,5-pyridinedicarboxylate, monobenzenesulphonate. Its empirical formula is C 20 H 25 ClN 2 O 5 ·C 6 H 6 O 3 S.
The atorvastatin calcium component of CADUET is chemically described as [R-(R*, R*)]-2-(4-fluorophenyl)-(beta), [dgr ]-di-hydroxy-5-(1-methylethyl)-3-phenyl-4-[(phenylamino)car-bonyl]-1H-pyrrole-1-heptanoic acid, calcium salt (2:1) trihydrate. Its empirical formula is (C 33 H 34 FN 2 O 5 ) 2 Ca·3H 2 O.
The structural formulae for amlodipine besylate and atorvastatin calcium are shown below.
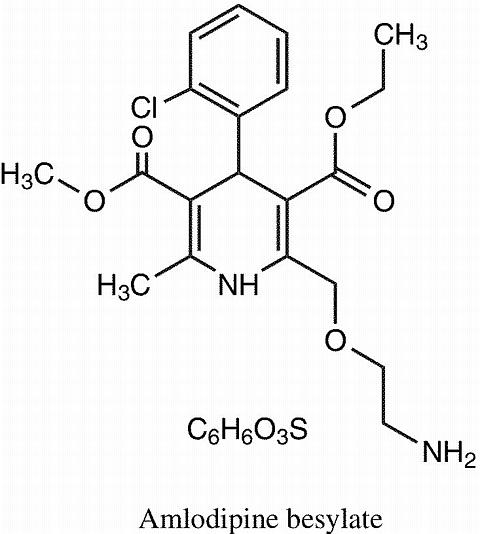
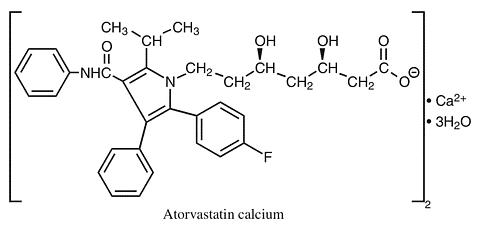
CADUET contains amlodipine besylate, a white to off-white crystalline powder, and atorvastatin calcium, also a white to off-white crystalline powder. Amlodipine besylate has a molecular weight of 567.1 and atorvastatin calcium has a molecular weight of 1209.42. Amlodipine besylate is slightly soluble in water and sparingly soluble in ethanol. Atorvastatin calcium is insoluble in aqueous solutions of pH 4 and below. Atorvastatin calcium is very slightly soluble in distilled water, pH 7.4 phosphate buffer, and acetonitrile; slightly soluble in ethanol, and freely soluble in methanol.
CADUET tablets are formulated for oral administration in the following strength combinations:
Table 1. CADUET Tablet Strengths2.5 mg/
10 mg2.5 mg/
20 mg2.5 mg/
40 mg5 mg/
10 mg5 mg/
20 mg5 mg/
40 mg5 mg/
80 mg10 mg/
10 mg10 mg/
20 mg10 mg/
40 mg10 mg/
80 mgamlodipine equivalent (mg) 2.5 2.5 2.5 5 5 5 5 10 10 10 10 atorvastatin equivalent (mg) 10 20 40 10 20 40 80 10 20 40 80
Each tablet also contains calcium carbonate, croscarmellose sodium, microcrystalline cellulose, pregelatinized starch, polysorbate 80, hydroxypropyl cellulose, purified water, colloidal silicon dioxide (anhydrous), magnesium stearate, Opadry ® II White 85F28751 (polyvinyl alcohol, titanium dioxide, PEG 3000 and talc) or Opadry ® II Blue 85F10919 (polyvinyl alcohol, titanium dioxide, PEG 3000, talc and FD&C blue #2). Combinations of atorvastatin with 2.5 mg and 5 mg amlodipine are film coated white, and combinations of atorvastatin with 10 mg amlodipine are film coated blue.
CLINICAL PHARMACOLOGY
Mechanism of Action
CADUET
CADUET is a combination of two drugs, a dihydropyridine calcium antagonist (calcium ion antagonist or slow-channel blocker) amlodipine (antihypertensive/antianginal agent) and an HMG-CoA reductase inhibitor atorvastatin (cholesterol lowering agent). The amlodipine component of CADUET inhibits the transmembrane influx of calcium ions into vascular smooth muscle and cardiac muscle. The atorvastatin component of CADUET is a selective, competitive inhibitor of HMG-CoA reductase, the rate-limiting enzyme that converts 3-hydroxy-3-methylglutaryl-coenzyme A to mevalonate, a precursor of sterols, including cholesterol.
The Amlodipine Component of CADUET
Experimental data suggest that amlodipine binds to both dihydropyridine and nondihydropyridine binding sites. The contractile processes of cardiac muscle and vascular smooth muscle are dependent upon the movement of extracellular calcium ions into these cells through specific ion channels. Amlodipine inhibits calcium ion influx across cell membranes selectively, with a greater effect on vascular smooth muscle cells than on cardiac muscle cells. Negative inotropic effects can be detected in vitro but such effects have not been seen in intact animals at therapeutic doses. Serum calcium concentration is not affected by amlodipine. Within the physiologic pH range, amlodipine is an ionized compound (pKa=8.6), and its kinetic interaction with the calcium channel receptor is characterized by a gradual rate of association and dissociation with the receptor binding site, resulting in a gradual onset of effect.
Amlodipine is a peripheral arterial vasodilator that acts directly on vascular smooth muscle to cause a reduction in peripheral vascular resistance and reduction in blood pressure.
The precise mechanisms by which amlodipine relieves angina have not been fully delineated, but are thought to include the following:
Exertional Angina: In patients with exertional angina, amlodipine reduces the total peripheral resistance (afterload) against which the heart works and reduces the rate pressure product, and thus myocardial oxygen demand, at any given level of exercise.
Vasospastic Angina: Amlodipine has been demonstrated to block constriction and restore blood flow in coronary arteries and arterioles in response to calcium, potassium epinephrine, serotonin, and thromboxane A 2 analog in experimental animal models and in human coronary vessels in vitro . This inhibition of coronary spasm is responsible for the effectiveness of amlodipine in vasospastic (Prinzmetal's or variant) angina.
The Atorvastatin Component of CADUET
Cholesterol and triglycerides circulate in the bloodstream as part of lipoprotein complexes. With ultracentrifugation, these complexes separate into HDL (high-density lipoprotein), IDL (intermediate-density lipoprotein), LDL (low-density lipoprotein), and VLDL (very-low-density lipoprotein) fractions. Triglycerides (TG) and cholesterol in the liver are incorporated into VLDL and released into the plasma for delivery to peripheral tissues. LDL is formed from VLDL and is catabolized primarily through the high-affinity LDL receptor.
Clinical and pathologic studies show that elevated plasma levels of total cholesterol (total-C), LDL-cholesterol (LDL-C), and apolipoprotein B (apo B) promote human atherosclerosis and are risk factors for developing cardiovascular disease, while increased levels of HDL-C are associated with a decreased cardiovascular risk.
Epidemiologic investigations have established that cardiovascular morbidity and mortality vary directly with the level of total-C and LDL-C, and inversely with the level of HDL-C.
In animal models, atorvastatin lowers plasma cholesterol and lipoprotein levels by inhibiting HMG-CoA reductase and cholesterol synthesis in the liver and by increasing the number of hepatic LDL receptors on the cell-surface to enhance uptake and catabolism of LDL; atorvastatin also reduces LDL production and the number of LDL particles.
Atorvastatin reduces total-C, LDL-C, and apo B in patients with homozygous and heterozygous familial hypercholesterolemia (FH), nonfamilial forms of hypercholesterolemia, and mixed dyslipidemia. Atorvastatin also reduces VLDL-C and TG and produces variable increases in HDL-C and apolipoprotein A-1. Atorvastatin reduces total-C, LDL-C, VLDL-C, apo B, TG, and non-HDL-C, and increases HDL-C in patients with isolated hypertriglyceridemia. Atorvastatin reduces intermediate density lipoprotein cholesterol (IDL-C) in patients with dysbetalipoproteinemia.
Like LDL, cholesterol-enriched triglyceride-rich lipoproteins, including VLDL, intermediate density lipoprotein (IDL), and remnants, can also promote atherosclerosis. Elevated plasma triglycerides are frequently found in a triad with low HDL-C levels and small LDL particles, as well as in association with non-lipid metabolic risk factors for coronary heart disease. As such, total plasma TG has not consistently been shown to be an independent risk factor for CHD. Furthermore, the independent effect of raising HDL or lowering TG on the risk of coronary and cardiovascular morbidity and mortality has not been determined.
Pharmacokinetics and Metabolism
Absorption
Studies with amlodipine: After oral administration of therapeutic doses of amlodipine alone, absorption produces peak plasma concentrations between 6 and 12 hours. Absolute bioavailability has been estimated to be between 64% and 90%. The bioavailability of amlodipine when administered alone is not altered by the presence of food.
Studies with atorvastatin: After oral administration alone, atorvastatin is rapidly absorbed; maximum plasma concentrations occur within 1 to 2 hours. Extent of absorption increases in proportion to atorvastatin dose. The absolute bioavailability of atorvastatin (parent drug) is approximately 14% and the systemic availability of HMG-CoA reductase inhibitory activity is approximately 30%. The low systemic availability is attributed to presystemic clearance in gastrointestinal mucosa and/or hepatic first-pass metabolism. Although food decreases the rate and extent of drug absorption by approximately 25% and 9%, respectively, as assessed by Cmax and AUC, LDL-C reduction is similar whether atorvastatin is given with or without food. Plasma atorvastatin concentrations are lower (approximately 30% for Cmax and AUC) following evening drug administration compared with morning. However, LDL-C reduction is the same regardless of the time of day of drug administration (see DOSAGE AND ADMINISTRATION ).
Studies with CADUET: Following oral administration of CADUET peak plasma concentrations of amlodipine and atorvastatin are seen at 6 to 12 hours and 1 to 2 hours post dosing, respectively. The rate and extent of absorption (bioavailability) of amlodipine and atorvastatin from CADUET are not significantly different from the bioavailability of amlodipine and atorvastatin administered separately (see above).
The bioavailability of amlodipine from CADUET was not affected by food. Although food decreases the rate and extent of absorption of atorvastatin from CADUET by approximately 32% and 11%, respectively, as it does with atorvastatin when given alone. LDL-C reduction is similar whether atorvastatin is given with or without food.
Distribution
Studies with amlodipine: Ex vivo studies have shown that approximately 93% of the circulating amlodipine drug is bound to plasma proteins in hypertensive patients. Steady-state plasma levels of amlodipine are reached after 7 to 8 days of consecutive daily dosing.
Studies with atorvastatin : Mean volume of distribution of atorvastatin is approximately 381 liters. Atorvastatin is >/=98% bound to plasma proteins. A blood/plasma ratio of approximately 0.25 indicates poor drug penetration into red blood cells. Based on observations in rats, atorvastatin calcium is likely to be secreted in human milk (see CONTRAINDICATIONS , Pregnancy and Lactation , and PRECAUTIONS , Nursing Mothers ).
Metabolism
Studies with amlodipine: Amlodipine is extensively (about 90%) converted to inactive metabolites via hepatic metabolism.
Studies with atorvastatin: Atorvastatin is extensively metabolized to ortho- and parahydroxylated derivatives and various beta-oxidation products. In vitro inhibition of HMG-CoA reductase by ortho- and parahydroxylated metabolites is equivalent to that of atorvastatin. Approximately 70% of circulating inhibitory activity for HMG-CoA reductase is attributed to active metabolites. In vitro studies suggest the importance of atorvastatin metabolism by cytochrome P450 3A4, consistent with increased plasma concentrations of atorvastatin in humans following coadministration with erythromycin, a known inhibitor of this isozyme (see PRECAUTIONS , Drug Interactions ). In animals, the ortho-hydroxy metabolite undergoes further glucuronidation.
Excretion
Studies with amlodipine: Elimination from the plasma is biphasic with a terminal elimination half-life of about 30-50 hours. Ten percent of the parent amlodipine compound and 60% of the metabolites of amlodipine are excreted in the urine.
Studies with atorvastatin: Atorvastatin and its me-tabolites are eliminated primarily in bile following he-patic and/or extra-hepatic metabolism; however, the drug does not appear to undergo enterohepatic recirculation. Mean plasma elimination half-life of atorvastatin in humans is approximately 14 hours, but the half-life of inhibitory activity for HMG-CoA reductase is 20 to 30 hours due to the contribution of active metabolites. Less than 2% of a dose of atorvastatin is recovered in urine following oral administration.
Special Populations
Geriatric
Studies with amlodipine: Elderly patients have decreased clearance of amlodipine with a resulting increase in AUC of approximately 40-60%, and a lower initial dose of amlodipine may be required.
Studies with atorvastatin: Plasma concentrations of atorvastatin are higher (approximately 40% for Cmax and 30% for AUC) in healthy elderly subjects (age >/=65 years) than in young adults. Clinical data suggest a greater degree of LDL-lowering at any dose of atorvastatin in the elderly population compared to younger adults (see PRECAUTIONS section, Geriatric Use ).
Pediatric
Studies with amlodipine: Sixty-two hypertensive patients aged 6 to 17 years received doses of amlodipine between 1.25 mg and 20 mg. Weight-adjusted clearance and volume of distribution were similar to values in adults.
Studies with atorvastatin: Pharmacokinetic data in the pediatric population are not available.
Gender
Studies with atorvastatin: Plasma concentrations of atorvastatin in women differ from those in men (approximately 20% higher for Cmax and 10% lower for AUC); however, there is no clinically significant difference in LDL-C reduction with atorvastatin between men and women.
Renal Insufficiency
Studies with amlodipine: The pharmacokinetics of amlodipine are not significantly influenced by renal impairment. Patients with renal failure may therefore receive the usual initial amlodipine dose.
Studies with atorvastatin: Renal disease has no influence on the plasma concentrations or LDL-C reduction of atorvastatin; thus, dose adjustment of atorvastatin in patients with renal dysfunction is not necessary (see DOSAGE AND ADMINISTRATION ).
Hemodialysis
While studies have not been conducted in patients with end-stage renal disease, hemodialysis is not expected to significantly enhance clearance of atorvastatin and/or amlodipine since both drugs are extensively bound to plasma proteins.
Hepatic Insufficiency
Studies with amlodipine: Elderly patients and patients with hepatic insufficiency have decreased clearance of amlodipine with a resulting increase in AUC of approximately 40-60%, and a lower initial dose may be required.
Studies with atorvastatin: In patients with chronic alcoholic liver disease, plasma concentrations of atorvastatin are markedly increased. Cmax and AUC are each 4-fold greater in patients with Childs-Pugh A disease. Cmax and AUC of atorvastatin are approximately 16-fold and 11-fold increased, respectively, in patients with Childs-Pugh B disease (see CONTRAINDICATIONS ).
Heart Failure
Studies with amlodipine: In patients with moderate to severe heart failure, the increase in AUC for amlodipine was similar to that seen in the elderly and in patients with hepatic insufficiency.
Pharmacodynamics
Hemodynamic Effects of Amlodipine: Following administration of therapeutic doses to patients with hypertension, amlodipine produces vasodilation resulting in a reduction of supine and standing blood pressures. These decreases in blood pressure are not accompanied by a significant change in heart rate or plasma catecholamine levels with chronic dosing. Although the acute intravenous administration of amlodipine decreases arterial blood pressure and increases heart rate in hemodynamic studies of patients with chronic stable angina, chronic administration of oral amlodipine in clinical trials did not lead to clinically significant changes in heart rate or blood pressures in normotensive patients with angina.
With chronic once daily oral administration of amlodipine, antihypertensive effectiveness is maintained for at least 24 hours. Plasma concentrations correlate with effect in both young and elderly patients. The magnitude of reduction in blood pressure with amlodipine is also correlated with the height of pretreatment elevation; thus, individuals with moderate hypertension (diastolic pressure 105-114 mmHg) had about a 50% greater response than patients with mild hypertension (diastolic pressure 90-104 mmHg). Normotensive subjects experienced no clinically significant change in blood pressures (+1/-2 mmHg).
In hypertensive patients with normal renal function, therapeutic doses of amlodipine resulted in a decrease in renal vascular resistance and an increase in glomerular filtration rate and effective renal plasma flow without change in filtration fraction or proteinuria.
As with other calcium channel blockers, hemodynamic measurements of cardiac function at rest and during exercise (or pacing) in patients with normal ventricular function treated with amlodipine have generally demonstrated a small increase in cardiac index without significant influence on dP/dt or on left ventricular end diastolic pressure or volume. In hemodynamic studies, amlodipine has not been associated with a negative inotropic effect when administered in the therapeutic dose range to intact animals and man, even when co-administered with beta-blockers to man. Similar findings, however, have been observed in normals or well-compensated patients with heart failure with agents possessing significant negative inotropic effects.
Electrophysiologic Effects of Amlodipine: Amlodipine does not change sinoatrial nodal function or atrioventricular conduction in intact animals or man. In patients with chronic stable angina, intravenous administration of 10 mg did not significantly alter A-H and H-V conduction and sinus node recovery time after pacing. Similar results were obtained in patients receiving amlodipine and concomitant beta blockers. In clinical studies in which amlodipine was administered in combination with beta-blockers to patients with either hypertension or angina, no adverse effects on electrocardiographic parameters were observed. In clinical trials with angina patients alone, amlodipine therapy did not alter electrocardiographic intervals or produce higher degrees of AV blocks.
LDL-C Reduction with Atorvastatin: Atorvastatin as well as some of its metabolites are pharmacologically active in humans. The liver is the primary site of action and the principal site of cholesterol synthesis and LDL clearance. Drug dosage rather than systemic drug concentration correlates better with LDL-C reduction. Individualization of drug dosage should be based on therapeutic response (see DOSAGE AND ADMINISTRATION ).
Clinical Studies
Clinical Studies with Amlodipine
Amlodipine Effects in Hypertension
Adult Patients: The antihypertensive efficacy of amlodipine has been demonstrated in a total of 15 double-blind, placebo-controlled, randomized studies involving 800 patients on amlodipine and 538 on placebo. Once daily administration produced statistically significant placebo-corrected reductions in supine and standing blood pressures at 24 hours postdose, averaging about 12/6 mmHg in the standing position and 13/7 mmHg in the supine position in patients with mild to moderate hypertension. Maintenance of the blood pressure effect over the 24-hour dosing interval was observed, with little difference in peak and trough effect. Tolerance was not demonstrated in patients studied for up to 1 year. The 3 parallel, fixed doses, dose response studies showed that the reduction in supine and standing blood pressures was dose-related within the recommended dosing range. Effects on diastolic pressure were similar in young and older patients. The effect on systolic pressure was greater in older patients, perhaps because of greater baseline systolic pressure. Effects were similar in black patients and in white patients.
Pediatric Patients: Two-hundred sixty-eight hypertensive patients aged 6 to 17 years were randomized first to amlodipine 2.5 or 5 mg once daily for 4 weeks and then randomized again to the same dose or to placebo for another 4 weeks. Patients receiving 5 mg amlodipine at the end of 8 weeks had lower blood pressure than those secondarily randomized to placebo. The magnitude of the treatment effect is difficult to interpret, but it is probably less than 5 mmHg systolic on the 5 mg dose. Adverse events were similar to those seen in adults.
Amlodipine Effects in Chronic Stable Angina: The effectiveness of 5-10 mg/day of amlodipine in exercise-induced angina has been evaluated in 8 placebo-controlled, double-blind clinical trials of up to 6 weeks duration involving 1038 patients (684 amlodipine, 354 placebo) with chronic stable angina. In 5 of the 8 studies, significant increases in exercise time (bicycle or treadmill) were seen with the 10 mg dose. Increases in symptom-limited exercise time averaged 12.8% (63 sec) for amlodipine 10 mg, and averaged 7.9% (38 sec) for amlodipine 5 mg. Amlodipine 10 mg also increased time to 1 mm ST segment deviation in several studies and decreased angina attack rate. The sustained efficacy of amlodipine in angina patients has been demonstrated over long-term dosing. In patients with angina, there were no clinically significant reductions in blood pressures (4/1 mmHg) or changes in heart rate (+0.3 bpm).
Amlodipine Effects in Vasospastic Angina: In a double-blind, placebo-controlled clinical trial of 4 weeks duration in 50 patients, amlodipine therapy decreased attacks by approximately 4/week compared with a placebo decrease of approximately 1/week (p<0.01). Two of 23 amlodipine and 7 of 27 placebo patients discontinued from the study due to lack of clinical improvement.
Amlodipine Effects in Patients with Congestive Heart Failure: Amlodipine has been compared to placebo in four 8-12 week studies of patients with NYHA class II/III heart failure, involving a total of 697 patients. In these studies, there was no evidence of worsened heart failure based on measures of exercise tolerance, NYHA classification, symptoms, or LVEF. In a long-term (follow-up at least 6 months, mean 13.8 months) placebo-controlled mortality/morbidity study of amlodipine 5-10 mg in 1153 patients with NYHA classes III (n=931) or IV (n=222) heart failure on stable doses of diuretics, digoxin, and ACE inhibitors, amlodipine had no effect on the primary endpoint of the study which was the combined endpoint of all-cause mortality and cardiac morbidity (as defined by life-threatening arrhythmia, acute myocardial infarction, or hospitalization for worsened heart failure), or on NYHA classification, or symptoms of heart failure. Total combined all-cause mortality and cardiac morbidity events were 222/571 (39%) for patients on amlodipine and 246/583 (42%) for patients on placebo; the cardiac morbid events represented about 25% of the endpoints in the study.
Another study (PRAISE-2) randomized patients with NYHA class III (80%) or IV (20%) heart failure without clinical symptoms or objective evidence of underlying ischemic disease, on stable doses of ACE inhibitor (99%), digitalis (99%) and diuretics (99%), to placebo (n=827) or amlodipine (n=827) and followed them for a mean of 33 months. There was no statistically significant difference between amlodipine and placebo in the primary endpoint of all cause mortality (95% confidence limits from 8% reduction to 29% increase on amlodipine). With amlodipine there were more reports of pulmonary edema.
Clinical Studies with Atorvastatin
Prevention of Cardiovascular Disease: In the Anglo-Scandinavian Cardiac Outcomes Trial (ASCOT), the effect of atorvastatin on fatal and non-fatal coronary heart disease was assessed in 10,305 hypertensive patients 40-80 years of age (mean of 63 years), without a previous myocardial infarction and with TC levels </=251 mg/dl (6.5 mmol/l). Additionally all patients had at least 3 of the following cardiovascular risk factors: male gender (81.1%), age >55 years (84.5%), smoking (33.2%), diabetes (24.3%), history of CHD in a first-degree relative (26%), TC:HDL >6 (14.3%), peripheral vascular disease (5.1%), left ventricular hypertrophy (14.4%), prior cerebrovascular event (9.8%), specific ECG abnormality (14.3%), proteinuria/albuminuria (62.4%)]. In this double-blind, placebo-controlled study patients were treated with anti-hypertensive therapy (Goal BP <140/90 mm Hg for non-diabetic patients, <130/80 mm Hg for diabetic patients) and allocated to either atorvastatin 10 mg daily (n=5168) or placebo (n=5137), using a covariate adaptive method which took into account the distribution of nine baseline characteristics of patients already enrolled and minimized the imbalance of those characteristics across the groups. Patients were followed for a median duration of 3.3 years.
The effect of 10 mg/day of atorvastatin on lipid levels was similar to that seen in previous clinical trials.
Atorvastatin significantly reduced the rate of coronary events [either fatal coronary heart disease (46 events in the placebo group vs 40 events in the atorvastatin group) or nonfatal MI (108 events in the placebo group vs 60 events in the atorvastatin group)] with a relative risk reduction of 36% [(based on incidences of 1.9% for atorvastatin vs 3.0% for placebo), p=0.0005 (see Figure 1)]. The risk reduction was consistent regardless of age, smoking status, obesity or presence of renal dysfunction. The effect of atorvastatin was seen regardless of baseline LDL levels. Due to the small number of events, results for women were inconclusive.
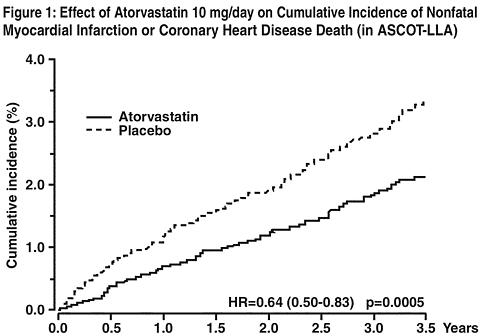
Atorvastatin also significantly decreased the relative risk for revascularization procedures by 42% Although the reduction of fatal and non-fatal strokes did not reach a pre-defined significance level (p=0.01), a favorable trend was observed with a 26% relative risk reduction (incidences of 1.7% for atorvastatin and 2.3% for placebo). There was no significant difference between the treatment groups for death due to cardiovascular causes (p=0.51) or noncardiovascular causes (p=0.17).
Atorvastatin Studies in Hypercholesterolemia (Heterozygous Familial and Nonfamilial) and Mixed Dyslipidemia (Fred-rickson Types IIa and IIb ) : Atorvastatin reduces total-C, LDL-C, VLDL-C, apo B, and TG, and increases HDL-C in patients with hypercholesterolemia and mixed dyslipidemia. Therapeutic response is seen within 2 weeks, and maximum response is usually achieved within 4 weeks and maintained during chronic therapy.
Atorvastatin is effective in a wide variety of patient populations with hypercholesterolemia, with and without hypertriglyceridemia, in men and women, and in the elderly.
In two multicenter, placebo-controlled, dose-response studies in patients with hypercholesterolemia, atorvastatin given as a single dose over 6 weeks significantly reduced total-C, LDL-C, apo B, and TG (pooled results are provided in Table 2).
Table 2. Dose-Response in Patients With Primary Hypercholesterolemia
(Adjusted Mean Percent Change From Baseline) aDOSE N TC LDL-C ApoB TG HDL-C Non-HDL-C/HDL-C Placebo 21 4 4 3 10 -3 7 10 mg22 -29 -39 -32 -19 6 -34 20 mg20 -33 -43 -35 -26 9 -41 40 mg21 -37 -50 -42 -29 6 -45 80 mg23 -45 -60 -50 -37 5 -53 a Results are pooled from 2 dose-response studies.
In patients with Fredrickson Types IIa and IIb hyperlipoproteinemia pooled from 24 controlled trials, the median (25 th and 75 th percentile) percent changes from baseline in HDL-C for atorvastatin 10, 20, 40, and 80 mg were 6.4 (-1.4, 14), 8.7 (0, 17), 7.8 (0, 16), and 5.1 (-2.7, 15), respectively. Additionally, analysis of the pooled data demonstrated consistent and significant decreases in total-C, LDL-C, TG, total-C/HDL-C, and LDL-C/HDL-C.
In three multicenter, double-blind studies in patients with hypercholesterolemia, atorvastatin was compared to other HMG-CoA reductase inhibitors. After randomization, patients were treated for 16 weeks with either atorvastatin 10 mg per day or a fixed dose of the comparative agent (Table 3).
Table 3. Mean Percent Change From Baseline at Endpoint
(Double-Blind, Randomized, Active-Controlled Trials)Treatment
(Daily Dose)N Total-C LDL-C Apo B TG HDL-C Non-HDL-C/
HDL-CStudy 1Atorvastatin 10 mg707 -27 a -36 a -28 a -17 a +7 -37 a Lovastatin 20 mg191 -19 -27 -20 -6 +7 -28 95% CI for Diff 1-9.2, -6.5 -10.7, -7.1 -10.0, -6.5 -15.2, -7.1 -1.7, 2.0 -11.1, -7.1 Study 2Atorvastatin 10 mg222 -25 b -35 b -27 b -17 b +6 -36 b Pravastatin 20 mg77 -17 -23 -17 -9 +8 -28 95% CI for Diff 1-10.8, -6.1 -14.5, -8.2 -13.4, -7.4 -14.1, -0.7 -4.9, 1.6 -11.5, -4.1 Study 3Atorvastatin 10 mg132 -29 c -37 c -34 c -23 c +7 -39 c Simvastatin 10 mg45 -24 -30 -30 -15 +7 -33 95% CI for Diff 1-8.7, -2.7 -10.1, -2.6 -8.0, -1.1 -15.1, -0.7 -4.3, 3.9 -9.6, -1.9 1 A negative value for the 95% CI for the difference between treatments favors atorvastatin for all except HDL-C, for which a positive value favors atorvastatin. If the range does not include 0, this indicates a statistically significant difference.a Significantly different from lovastatin, ANCOVA, p </=0.05b Significantly different from pravastatin, ANCOVA, p </=0.05c Significantly different from simvastatin, ANCOVA, p </=0.05
The impact on clinical outcomes of the differences in lipid-altering effects between treatments shown in Table 3 is not known. Table 3 does not contain data comparing the effects of atorvastatin 10 mg and higher doses of lovastatin, pravastatin, and simvastatin. The drugs compared in the studies summarized in the table are not necessarily interchangeable.
Atorvastatin Effects in Hypertriglyceridemia (Fredrickson Type IV ) : The response to atorvastatin in 64 patients with isolated hypertriglyceridemia treated across several clinical trials is shown in the table below. For the atorvastatin-treated patients, median (min, max) baseline TG level was 565 (267-1502).
Table 4. Combined Patients With Isolated Elevated TG: Median (min, max) Percent Changes From BaselinePlacebo
(N=12)Atorvastatin 10 mg
(N=37)Atorvastatin 20 mg
(N=13)Atorvastatin 80 mg
(N=14)Triglycerides-12.4 (-36.6, 82.7) -41.0 (-76.2, 49.4) -38.7 (-62.7, 29.5) -51.8 (-82.8, 41.3) Total-C-2.3 (-15.4, 24.4) -28.2 (-44.9, -6.8) -34.9 (-49.6, -15.2) -44.4 (-63.5, -3.8) LDL-C3.6 (-31.3, 31.6) -26.5 (-57.7, 9.8) -30.4 (-53.9, 0.3) -40.5 (-60.6, -13.8) HDL-C3.8 (-18.6, 13.4) 13.8 (-9.7, 61.5) 11.0 (-3.2, 25.2) 7.5 (-10.8, 37.2) VLDL-C-1.0 (-31.9, 53.2) -48.8 (-85.8, 57.3) -44.6 (-62.2, -10.8) -62.0 (-88.2, 37.6) non-HDL-C-2.8 (-17.6, 30.0) -33.0 (-52.1, -13.3) -42.7 (-53.7, -17.4) -51.5 (-72.9, -4.3)
Atorvastatin Effects in Dysbetalipoproteinemia (Fredrickson Type III ) : The results of an open-label crossover study of atorvastatin in 16 patients (genotypes: 14 apo E2/E2 and 2 apo E3/E2) with dysbetalipoproteinemia ( Fredrickson Type III) are shown in the table below.
Table 5. Open-Label Crossover Study of 16 Patients With Dysbetalipoproteinemia ( Fredrickson Type III)Median (min, max)
at Baseline (mg/dL)Median % Change (min, max) Atorvastatin 10 mg Atorvastatin 80 mg Total-C442 (225, 1320) -37 (-85, 17) -58 (-90, -31) Triglycerides678 (273, 5990) -39 (-92, -8) -53 (-95, -30) IDL-C + VLDL-C215 (111, 613) -32 (-76, 9) -63 (-90, -8) non-HDL-C411 (218, 1272) -43 (-87, -19) -64 (-92, -36)
Atorvastatin Effects in Homozygous Familial Hypercholesterolemia: In a study without a concurrent control group, 29 patients ages 6 to 37 years with homozygous FH received maximum daily doses of 20 to 80 mg of atorvastatin. The mean LDL-C reduction in this study was 18%. Twenty-five patients with a reduction in LDL-C had a mean response of 20% (range of 7% to 53%, median of 24%); the remaining 4 patients had 7% to 24% increases in LDL-C. Five of the 29 patients had absent LDL-receptor function. Of these, 2 patients also had a portacaval shunt and had no significant reduction in LDL-C. The remaining 3 receptor-negative patients had a mean LDL-C reduction of 22%.
Atorvastatin Effects in Heterozygous Familial Hypercholesterolemic Pediatric Patients: In a double-blind, placebo-controlled study followed by an open-label phase, 187 boys and postmenarchal girls 10-17 years of age (mean age 14.1 years) with heterozygous FH or severe hypercholesterolemia were randomized to atorvastatin (n=140) or placebo (n=47) for 26 weeks and then all received atorvastatin for 26 weeks. Inclusion in the study required 1) a baseline LDL-C level >/= 190 mg/dL or 2) a baseline LDL-C >/= 160 mg/dL and positive family history of FH or documented premature cardiovascular disease in a first- or second-degree relative. The mean baseline LDL-C value was 218.6 mg/dL (range: 138.5-385.0 mg/dL) in the atorvastatin group compared to 230.0 mg/dL (range: 160.0-324.5 mg/dL) in placebo group. The dosage of atorvastatin (once daily) was 10 mg for the first 4 weeks and up-titrated to 20 mg if the LDL-C level was > 130 mg/dL. The number of atorvastatin-treated patients who required up-titration to 20 mg after Week 4 during the double-blind phase was 80 (57.1%).
Atorvastatin significantly decreased plasma levels of total-C, LDL-C, triglycerides, and apolipoprotein B during the 26 week double-blind phase (see Table 6).
Table 6. Lipid-altering Effects of Atorvastatin in Adolescent Boys and Girls with Heterozygous Familial Hypercholesterolemia or Severe Hypercholesterolemia (Mean Percent Change from Baseline at Endpoint in Intention-to-Treat Population)DOSAGE N Total-C LDL-C HDL-C TG Apolipoprotein B Placebo 47 -1.5 -0.4 -1.9 1.0 0.7 Atorvastatin 140 -31.4 -39.6 2.8 -12.0 -34.0
The mean achieved LDL-C value was 130.7 mg/dL (range: 70.0-242.0 mg/dL) in the atorvastatin group compared to 228.5 mg/dL (range: 152.0-385.0 mg/dL) in the placebo group during the 26 week double-blind phase.
The safety and efficacy of atorvastatin doses above 20 mg have not been studied in controlled trials in children. The long-term efficacy of atorvastatin therapy in childhood to reduce morbidity and mortality in adulthood has not been established.
Clinical Study of Combined Amlodipine and Atorvastatin in Patients with Hypertension and Dyslipidemia
In a double-blind, placebo-controlled study, a total of 1660 patients with co-morbid hypertension and dyslipidemia received once daily treatment with eight dose combinations of amlodipine and atorvastatin (5/10, 10/10, 5/20, 10/20, 5/40, 10/40, 5/80, or 10/80 mg), amlodipine alone (5 mg or 10 mg), atorvastatin alone (10 mg, 20 mg, 40 mg, or 80 mg) or placebo. In addition to concomitant hypertension and dyslipidemia, 15% of the patients had diabetes mellitus, 22% were smokers and 14% had a positive family history of cardiovascular disease. At eight weeks, all eight combination-treatment groups of amlodipine and atorvastatin demonstrated statistically significant dose-related reductions in systolic blood pressure (SBP), diastolic blood pressure (DBP) and LDL-C compared to placebo, with no overall modification of effect of either component on SBP, DBP and LDL-C (Table 7).
Table 7. Efficacy in Terms of Reduction in Blood Pressure and LDL-C Efficacy of the Combined Treatments in Reducing Systolic BP Parameter / Analysis ATO 0 mg ATO 10 mg ATO 20 mg ATO 40 mg ATO 80 mg AML 0 mgMean Change
(mmHg)-3.0 -4.5 -6.2 -6.2 -6.4 Difference versus
placebo (mmHg)- -1.5 -3.2 -3.2 -3.4 AML 5 mgMean change
(mmHg)-12.8 -13.7 -15.3 -12.7 -12.2 Difference versus
placebo (mmHg)-9.8 -10.7 -12.3 -9.7 -9.2 AML 10 mgMean change
(mmHg)-16.2 -15.9 -16.1 -16.3 -17.6 Difference versus
placebo (mmHg)-13.2 -12.9 -13.1 -13.3 -14.6 Efficacy of the Combined Treatments in Reducing Diastolic BPParameter / AnalysisATO 0 mg ATO 10 mg ATO 20 mg ATO 40 mg ATO 80 mg AML 0 mgMean change
(mmHg)-3.3 -4.1 -3.9 -5.1 -4.1 Difference versus
placebo (mmHg)- -0.8 -0.6 -1.8 -0.8 AML 5 mgMean change
(mmHg)-7.6 -8.2 -9.4 -7.3 -8.4 Difference versus
placebo (mmHg)-4.3 -4.9 -6.1 -4.0 -5.1 AML 10 mgMean change
(mmHg)-10.4 -9.1 -10.6 -9.8 -11.1 Difference versus
placebo (mmHg)-7.1 -5.8 -7.3 -6.5 -7.8 Efficacy of the Combined Treatments in Reducing LDL-C (% change)Parameter / AnalysisATO 0 mg ATO 10 mg ATO 20 mg ATO 40 mg ATO 80 mg AML 0 mgMean % change -1.1 -33.4 -39.5 -43.1 -47.2 AML 5 mgMean % change -0.1 -38.7 -42.3 -44.9 -48.4 AML 10 mgMean % change -2.5 -36.6 -38.6 -43.2 -49.1 INDICATIONS AND USAGE
CADUET (amlodipine and atorvastatin) is indicated in patients for whom treatment with both amlodipine and atorvastatin is appropriate.
Amlodipine
- Hypertension: Amlodipine is indicated for the treatment of hypertension. It may be used alone or in combination with other antihypertensive agents;
- Chronic Stable Angina: Amlodipine is indicated for the treatment of chronic stable angina. Amlodipine may be used alone or in combination with other antianginal or antihypertensive agents;
- Vasospastic Angina (Prinzmetal's or Variant Angina): Amlodipine is indicated for the treatment of confirmed or suspected vasospastic angina. Amlodipine may be used as monotherapy or in combination with other antianginal drugs.
AND
Atorvastatin
-
Prevention of Cardiovascular Disease:
In adult patients without clinically evident coronary heart disease, but with multiple risk factors for coronary heart disease such as age >/= 55 years, smoking, hypertension, low HDL-C, or a family history of early coronary heart disease, atorvastatin is indicated to:
- Reduce the risk of myocardial infarction
- Reduce the risk for revascularization procedures and angina
- Heterozygous Familial and Nonfamilial Hypercholesterolemia: Atorvastatin is indicated as an adjunct to diet to reduce elevated total-C, LDL-C, apo B, and TG levels and to increase HDL-C in patients with primary hypercholesterolemia (heterozygous familial and nonfamilial) and mixed dyslipidemia ( Fredrickson Types IIa and IIb);
- Elevated Serum TG Levels: Atorvastatin is indicated as an adjunct to diet for the treatment of patients with elevated serum TG levels ( Fredrickson Type IV);
- Primary Dysbetalipoproteinemia: Atorvastatin is indicated for the treatment of patients with primary dysbetalipoproteinemia ( Fredrickson Type III) who do not respond adequately to diet;
- Homozygous Familial Hypercholesterolemia: Atorvastatin is indicated to reduce total-C and LDL-C in patients with homozygous familial hypercholesterolemia as an adjunct to other lipid-lowering treatments (e.g., LDL apheresis) or if such treatments are unavailable;
-
Pediatric Patients:
Atorvastatin is indicated as an adjunct to diet to reduce total-C, LDL-C, and apo B levels in boys and postmenarchal girls, 10 to 17 years of age, with heterozygous familial hypercholesterolemia if after an adequate trial of diet therapy the following findings are present:
- LDL-C remains >/= 190 mg/dL or
-
LDL-C remains >/= 160 mg/dL and:
- there is a positive family history of premature cardiovascular disease or
- two or more other CVD risk factors are present in the pediatric patients.
Therapy with lipid-altering agents should be a component of multiple-risk-factor intervention in individuals at increased risk for atherosclerotic vascular disease due to hypercholesterolemia. Lipid-altering agents should be used, in addition to a diet restricted in saturated fat and cholesterol, only when the response to diet and other nonpharmacological measures has been inadequate (see National Cholesterol Education Program (NCEP) Guidelines, summarized in Table 8).
Table 8. NCEP Treatment Guidelines: LDL-C Goals and Cutpoints for Therapeutic Lifestyle Changes and Drug Therapy in Different Risk CategoriesRisk Category LDL-C Goal
(mg/dL)LDL-C Level at Which to
Initiate Therapeutic
Lifestyle Changes
(mg/dL)LDL-C Level at Which to
Consider
Drug
Therapy (mg/dL)CHD a or CHD risk
equivalents
(10-year risk >20%)<100 >/=100 >/=130
(100-129: drug optional) b2+ Risk Factors
(10-year risk </=20%)<130 >/=130 10-year risk 10%-20%: >/=130
10-year risk <10%: >/=1600-1 Risk Factor c <160 >/=160 >/=190
(160-189: LDL-lowering drug optional)a CHD, coronary heart disease b Some authorities recommend use of LDL-lowering drugs in this category if an LDL-C level of <100 mg/dL cannot be achieved by therapeutic lifestyle changes. Others prefer use of drugs that primarily modify triglycerides and HDL-C, e.g., nicotinic acid or fibrate. Clinical judgement also may call for deferring drug therapy in this subcategory. c Almost all people with 0-1 risk factor have 10-year risk <10%; thus, 10-year risk assessment in people with 0-1 risk factor is not necessary. After the LDL-C goal has been achieved, if the TG is still >/= 200 mg/dL, non-HDL-C (total-C minus HDL-C) becomes a secondary target of therapy. Non-HDL-C goals are set 30 mg/dL higher than LDL-C goals for each risk category.
Prior to initiating therapy with atorvastatin, secondary causes for hypercholesterolemia (e.g., poorly controlled diabetes mellitus, hypothyroidism, nephrotic syndrome, dysproteinemias, obstructive liver disease, other drug therapy, and alcoholism) should be excluded, and a lipid profile performed to measure total-C, LDL-C, HDL-C, and TG. For patients with TG <400 mg/dL (<4.5 mmol/L), LDL-C can be estimated using the following equation: LDL-C = total-C - (0.20 x [TG] + HDL-C). For TG levels >400 mg/dL (>4.5 mmol/L), this equation is less accurate and LDL-C concentrations should be determined by ultracentrifugation.
The antidyslipidemic component of CADUET has not been studied in conditions where the major lipoprotein abnormality is elevation of chylomicrons ( Fredrickson Types I and V).
The NCEP classification of cholesterol levels in pediatric patients with a familial history of hypercholesterolemia or premature cardiovascular disease is summarized below:
Table 9. NCEP Classification of Cholesterol Levels in Pediatric PatientsCategoryTotal-C (mg/dL) LDL-C (mg/dL) Acceptable<170 <110 Borderline170-199 110-129 High>/=200 >/=130 CONTRAINDICATIONS
CADUET contains atorvastatin and is therefore contraindicated in patients with active liver disease or unexplained persistent elevations of serum transaminases.
CADUET is contraindicated in patients with known hypersensitivity to any component of this medication.
Pregnancy and Lactation
Atherosclerosis is a chronic process and discontinuation of lipid-lowering drugs during pregnancy should have little impact on the outcome of long-term therapy of primary hypercholesterolemia. Cholesterol and other products of cholesterol biosynthesis are essential components for fetal development (including synthesis of steroids and cell membranes). Since HMG-CoA reductase inhibitors decrease cholesterol synthesis and possibly the synthesis of other biologically active substances derived from cholesterol, they may cause fetal harm when administered to pregnant women. Therefore, HMG-CoA reductase inhibitors are contraindicated during pregnancy and in nursing mothers. CADUET, WHICH INCLUDES ATORVASTATIN, SHOULD BE ADMINISTERED TO WOMEN OF CHILDBEARING AGE ONLY WHEN SUCH PATIENTS ARE HIGHLY UNLIKELY TO CONCEIVE AND HAVE BEEN INFORMED OF THE POTENTIAL HAZARDS. If the patient becomes pregnant while taking this drug, therapy should be discontinued and the patient apprised of the potential hazard to the fetus.
WARNINGS
Increased Angina and/or Myocardial Infarction
Rarely, patients, particularly those with severe obstructive coronary artery disease, have developed documented increased frequency, duration and/or severity of angina or acute myocardial infarction on starting calcium channel blocker therapy or at the time of dosage increase. The mechanism of this effect has not been elucidated.
Liver Dysfunction
HMG-CoA reductase inhibitors, like some other lipid-lowering therapies, have been associated with biochemical abnormalities of liver function. Persistent elevations (>3 times the upper limit of normal [ULN] occurring on 2 or more occasions) in serum transaminases occurred in 0.7% of patients who received atorvastatin in clinical trials. The incidence of these abnormalities was 0.2%, 0.2%, 0.6%, and 2.3% for 10, 20, 40, and 80 mg, respectively.
In clinical trials in patients taking atorvastatin the following has been observed. One patient in clinical trials developed jaundice. Increases in liver function tests (LFT) in other patients were not associated with jaundice or other clinical signs or symptoms. Upon dose reduction, drug interruption, or discontinuation, transaminase levels returned to or near pretreatment levels without sequelae. Eighteen of 30 patients, with persistent LFT elevations continued treatment with a reduced dose of atorvastatin.
It is recommended that liver function tests be performed prior to and at 12 weeks following both the initiation of therapy and any elevation of dose, and periodically (e.g., semiannually) thereafter . Liver enzyme changes generally occur in the first 3 months of treatment with atorvastatin. Patients who develop increased transaminase levels should be monitored until the abnormalities resolve. Should an increase in ALT or AST of >3 times ULN persist, reduction of dose or withdrawal of CADUET is recommended.
CADUET should be used with caution in patients who consume substantial quantities of alcohol and/or have a history of liver disease. Active liver disease or unexplained persistent transaminase elevations are contraindications to the use of CADUET (see CONTRAINDICATIONS ).
Skeletal Muscle
Rare cases of rhabdomyolysis with acute renal failure secondary to myoglobinuria have been reported with the atorvastatin component of CADUET and with other drugs in the HMG-CoA reductase inhibitor class.
Uncomplicated myalgia has been reported in atorvastatin-treated patients (see ADVERSE REACTIONS ). Myopathy, defined as muscle aches or muscle weakness in conjunction with increases in creatine phosphokinase (CPK) values >10 times ULN, should be considered in any patient with diffuse myalgias, muscle tenderness or weakness, and/or marked elevation of CPK. Patients should be advised to report promptly unexplained muscle pain, tenderness or weakness, particularly if accompanied by malaise or fever. CADUET therapy should be discontinued if markedly elevated CPK levels occur or myopathy is diagnosed or suspected.
The risk of myopathy during treatment with drugs in the HMG-CoA reductase inhibitor class is increased with concurrent administration of cyclosporine, fibric acid derivatives, erythromycin, niacin, or azole antifungals. Physicians considering combined therapy with CADUET and fibric acid derivatives, erythromycin, immunosuppressive drugs, azole antifungals, or lipid-lowering doses of niacin should carefully weigh the potential benefits and risks and should carefully monitor patients for any signs or symptoms of muscle pain, tenderness, or weakness, particularly during the initial months of therapy and during any periods of upward dosage titration of either drug. Periodic creatine phosphokinase (CPK) determinations may be considered in such situations, but there is no assurance that such monitoring will prevent the occurrence of severe myopathy.
In patients taking CADUET, therapy should be temporarily withheld or discontinued in any patient with an acute, serious condition suggestive of a myopathy or having a risk factor predisposing to the development of renal failure secondary to rhabdomyolysis (e.g., severe acute infection, hypotension, major surgery, trauma, severe metabolic, endocrine and electrolyte disorders, and uncontrolled seizures).
PRECAUTIONS
General
Since the vasodilation induced by the amlodipine component of CADUET is gradual in onset, acute hypotension has rarely been reported after oral administration of amlodipine. Nonetheless, caution should be exercised when administering CADUET as with any other peripheral vasodilator particularly in patients with severe aortic stenosis.
Before instituting therapy with CADUET, an attempt should be made to control hypercholesterolemia with appropriate diet, exercise, and weight reduction in obese patients, and to treat other underlying medical problems (see INDICATIONS AND USAGE ).
Use in Patients with Congestive Heart Failure
In general, calcium channel blockers should be used with caution in patients with heart failure. The amlodipine component of CADUET (5-10 mg per day) has been studied in a placebo-controlled trial of 1153 patients with NYHA Class III or IV heart failure (see CLINICAL PHARMACOLOGY ) on stable doses of ACE inhibitor, digoxin, and diuretics. Follow-up was at least 6 months, with a mean of about 14 months. There was no overall adverse effect on survival or cardiac morbidity (as defined by life-threatening arrhythmia, acute myocardial infarction, or hospitalization for worsened heart failure). Amlodipine has been compared to placebo in four 8-12 week studies of patients with NYHA class II/III heart failure, involving a total of 697 patients. In these studies, there was no evidence of worsened heart failure based on measures of exercise tolerance, NYHA classification, symptoms, or LVEF.
Beta-Blocker Withdrawal
The amlodipine component of CADUET is not a beta-blocker and therefore gives no protection against the dangers of abrupt beta-blocker withdrawal; any such withdrawal should be by gradual reduction of the dose of beta-blocker.
Endocrine Function
HMG-CoA reductase inhibitors, such as the atorvastatin component of CADUET interfere with cholesterol synthesis and theoretically might blunt adrenal and/or gonadal steroid production. Clinical studies have shown that atorvastatin does not reduce basal plasma cortisol concentration or impair adrenal reserve. The effects of HMG-CoA reductase inhibitors on male fertility have not been studied in adequate numbers of patients. The effects, if any, on the pituitary-gonadal axis in premenopausal women are unknown. Caution should be exercised if an HMG-CoA reductase inhibitor is administered concomitantly with drugs thatmay decrease the levels or activity of endogenous steroid hormones, such as ketoconazole, spironolactone, and cimetidine.
CNS Toxicity
Studies with atorvastatin: Brain hemorrhage was seen in a female dog treated with atorvastatin calcium for 3 months at a dose equivalent to 120 mg atorvastatin/kg/day. Brain hemorrhage and optic nerve vacuolation were seen in another female dog that was sacrificed in moribund condition after 11 weeks of escalating doses of atorvastatin calcium equivalent to up to 280 mg atorvastatin/kg/day. The 120 mg/kg dose of atorvastatin resulted in a systemic exposure approximately 16 times the human plasma area-under-the-curve (AUC, 0-24 hours) based on the maximum human dose of 80 mg/day. A single tonic convulsion was seen in each of 2 male dogs (one treated with atorvastatin calcium at a dose equivalent to 10 mg atorvastatin/kg/day and one at a dose equivalent to 120 mg atorvastatin/kg/day) in a 2-year study. No CNS lesions have been observed in mice after chronic treatment for up to 2 years at doses of atorvastatin calcium equivalent to up to 400 mg atorvastatin/kg/day or in rats at doses equivalent to up to 100 mg atorvastatin/kg/day. These doses were 6 to 11 times (mouse) and 8 to 16 times (rat) the human AUC (0-24) based on the maximum recommended human dose of 80 mg atorvastatin/day.
CNS vascular lesions, characterized by perivascular hemorrhages, edema, and mononuclear cell infiltration of perivascular spaces, have been observed in dogs treated with other members of the HMG-CoA reductase class. A chemically similar drug in this class produced optic nerve degeneration (Wallerian degeneration of retinogeniculate fibers) in clinically normal dogs in a dose-dependent fashion at a dose that produced plasma drug levels about 30 times higher than the mean drug level in humans taking the highest recommended dose.
Information for Patients
Due to the risk of myopathy with drugs of the HMG-CoA reductase class, to which the atorvastatin component of CADUET belongs, patients should be advised to report promptly unexplained muscle pain, tenderness, or weakness, particularly if accompanied by malaise or fever.
Drug Interactions
Data from a drug-drug interaction study involving 10 mg of amlodipine and 80 mg of atorvastatin in healthy subjects indicate that the pharmacokinetics of amlodipine are not altered when the drugs are coadministered. The effect of amlodipine on the pharmacokinetics of atorvastatin showed no effect on the Cmax: 91% (90% confidence interval: 80 to 103%), but the AUC of atorvastatin increased by 18% (90% confidence interval: 109 to 127%) in the presence of amlodipine.
No drug interaction studies have been conducted with CADUET and other drugs, although studies have been conducted in the individual amlodipine and atorvastatin components, as described below:
Studies with Amlodipine:
In vitro data in human plasma indicate that amlodipine has no effect on the protein binding of drugs tested (digoxin, phenytoin, warfarin, and indomethacin).
Cimetidine : Co-administration of amlodipine with cimetidine did not alter the pharmacokinetics of amlodipine.
Maalox ® (antacid): Co-administration of the antacid Maalox with a single dose of amlodipine had no significant effect on the pharmacokinetics of amlodipine.
Sildenafil : A single 100 mg dose of sildenafil (Viagra ® ) in subjects with essential hypertension had no effect on the pharmacokinetic parameters of amlodipine. When amlodipine and sildenafil were used in combination, each agent independently exerted its own blood pressure lowering effect.
Digoxin: Co-administration of amlodipine with digoxin did not change serum digoxin levels or digoxin renal clearance in normal volunteers.
Ethanol (alcohol): Single and multiple 10 mg doses of amlodipine had no significant effect on the pharmacokinetics of ethanol.
Warfarin: Co-administration of amlodipine with warfarin did not change the warfarin prothrombin response time.
In clinical trials, amlodipine has been safely administered with thiazide diuretics, beta-blockers, angiotensin-converting enzyme inhibitors, long-acting nitrates, sublingual nitroglycerin, digoxin, warfarin, non-steroidal anti-inflammatory drugs, antibiotics, and oral hypoglycemic drugs.
Studies with Atorvastatin:
The risk of myopathy during treatment with drugs of the HMG-CoA reductase class is increased with concurrent administration of cyclosporine, fibric acid derivatives, niacin (nicotinic acid), erythromycin, or azole antifungals (see WARNINGS , Skeletal Muscle ).
Antacid: When atorvastatin and Maalox TC suspension were coadministered, plasma concentrations of atorvastatin decreased approximately 35%. However, LDL-C reduction was not altered.
Antipyrine: Because atorvastatin does not affect the pharmacokinetics of antipyrine, interactions with other drugs metabolized via the same cytochrome isozymes are not expected.
Colestipol: Plasma concentrations of atorvastatin decreased approximately 25% when colestipol and atorvastatin were coadministered. However, LDL-C reduction was greater when atorvastatin and colestipol were coadministered than when either drug was given alone.
Cimetidine: Atorvastatin plasma concentrations and LDL-C reduction were not altered by coadministration of cimetidine.
Digoxin: When multiple doses of atorvastatin and digoxin were coadministered, steady-state plasma digoxin concentrations increased by approximately 20%. Patients taking digoxin should be monitored appropriately.
Erythromycin: In healthy individuals, plasma concentrations of atorvastatin increased approximately 40% with coadministration of atorvastatin and erythromycin, a known inhibitor of cytochrome P450 3A4 (see WARNINGS , Skeletal Muscle ).
Oral Contraceptives: Coadministration of atorvastatin and an oral contraceptive increased AUC values for norethindrone and ethinyl estradiol by approximately 30% and 20%. These increases should be considered when selecting an oral contraceptive for a woman taking CADUET.
Warfarin: Atorvastatin had no clinically significant effect on prothrombin time when administered to patients receiving chronic warfarin treatment.
Drug/Laboratory Test Interactions
None known.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Studies with amlodipine: Rats and mice treated with amlodipine maleate in the diet for up to two years, at concentrations calculated to provide daily dosage levels of 0.5, 1.25, and 2.5 mg amlodipine/kg/day, showed no evidence of a carcinogenic effect of the drug. For the mouse, the highest dose was, on a mg/m 2 basis, similar to the maximum recommended human dose of 10 mg amlodipine/day * . For the rat, the highest dose level was, on a mg/m 2 basis, about twice the maximum recommended human dose * .
Mutagenicity studies conducted with amlodipine maleate revealed no drug related effects at either the gene or chromosome levels.
There was no effect on the fertility of rats treated orally with amlodipine maleate (males for 64 days and females for 14 days prior to mating) at doses up to 10 mg amlodipine/kg/day (8 times * the maximum recommended human dose of 10 mg/day on a mg/m 2 basis).
Studies with atorvastatin: In a 2-year carcinogenicity study with atorvastatin calcium in rats at dose levels equivalent to 10, 30, and 100 mg atorvastatin/kg/day, 2 rare tumors were found in muscle in high-dose females: in one, there was a rhabdomyosarcoma and, in another, there was a fibrosarcoma. This dose represents a plasma AUC (0-24) value of approximately 16 times the mean human plasma drug exposure after an 80 mg oral dose.
A 2-year carcinogenicity study in mice given atorvastatin calcium at dose levels equivalent to 100, 200, and 400 mg atorvastatin/kg/day resulted in a significant increase in liver adenomas in high-dose males and liver carcinomas in high-dose females. These findings occurred at plasma AUC (0-24) values of approximately 6 times the mean human plasma drug exposure after an 80 mg oral dose.
In vitro , atorvastatin was not mutagenic or clastogenic in the following tests with and without metabolic activation: the Ames test with Salmonella typhimurium and Escherichia coli , the HGPRT forward mutation assay in Chinese hamster lung cells, and the chromosomal aberration assay in Chinese hamster lung cells. Atorvastatin was negative in the in vivo mouse micronucleus test.
There were no effects on fertility when rats were given atorvastatin calcium at doses equivalent to up to 175 mg atorvastatin/kg/day (15 times the human exposure). There was aplasia and aspermia in the epididymides of 2 of 10 rats treated with atorvastatin calcium at a dose equivalent to 100 mg atorvastatin/kg/day for 3 months (16 times the human AUC at the 80 mg dose); testis weights were significantly lower at 30 and 100 mg/kg/day and epididymal weight was lower at 100 mg/kg/day. Male rats given the equivalent of 100 mg atorvastatin/kg/day for 11 weeks prior to mating had decreased sperm motility, spermatid head concentration, and increased abnormal sperm. Atorvastatin caused no adverse effects on semen parameters, or reproductive organ histopathology in dogs given doses of atorvastatin calcium equivalent to 10, 40, or 120 mg atorvastatin/kg/day for two years.
Pregnancy
Pregnancy Category X (see CONTRAINDICATIONS )
Safety in pregnant women has not been established with CADUET. CADUET should be administered to women of child-bearing potential only when such patients are highly unlikely to conceive and have been informed of the potential hazards. If the woman becomes pregnant while taking CADUET, it should be discontinued and the patient advised again as to the potential hazards to the fetus.
Studies with amlodipine: No evidence of teratogenicity or other embryo/fetal toxicity was found when pregnant rats and rabbits were treated orally with amlodipine maleate at doses up to 10 mg amlodipine/kg/day (respectively 8 times * and 23 times * the maximum recommended human dose of 10 mg/day on a mg/m 2 basis) during their respective periods of major organogenesis. However, litter size was significantly decreased (by about 50%) and the number of intrauterine deaths was significantly increased (about 5-fold) in rats receiving amlodipine maleate at 10 mg amlodipine/kg/day for 14 days before mating and throughout mating and gestation. Amlodipine maleate has been shown to prolong both the gestation period and the duration of labor in rats at this dose. There are no adequate and well-controlled studies in pregnant women.
* Based on patient weight of 50 kg.
Studies with atorvastatin: Atorvastatin crosses the rat placenta and reaches a level in fetal liver equivalent to that of maternal plasma. Atorvastatin was not teratogenic in rats at doses of atorvastatin calcium equivalent to up to 300 mg atorvastatin/kg/day or in rabbits at doses of atorvastatin calcium equivalent to up to 100 mg atorvastatin/kg/day. These doses resulted in multiples of about 30 times (rat) or 20 times (rabbit) the human exposure based on surface area (mg/m 2 ).
In a study in rats given atorvastatin calcium at doses equivalent to 20, 100, or 225 mg atorvastatin/kg/day, from gestation day 7 through to lactation day 21 (weaning), there was decreased pup survival at birth, neonate, weaning, and maturity for pups of mothers dosed with 225 mg/kg/day. Body weight was decreased on days 4 and 21 for pups of mothers dosed at 100 mg/kg/day; pup body weight was decreased at birth and at days 4, 21, and 91 at 225 mg/kg/day. Pup development was delayed (rotorod performance at 100 mg/kg/day and acoustic startle at 225 mg/kg/day; pinnae detachment and eye opening at 225 mg/kg/day). These doses of atorvastatin correspond to 6 times (100 mg/kg) and 22 times (225 mg/kg) the human AUC at 80 mg/day.
Rare reports of congenital anomalies have been received following intrauterine exposure to HMG-CoA reductase inhibitors. There has been one report of severe congenital bony deformity, tracheo-esophageal fistula, and anal atresia (VATER association) in a baby born to a woman who took lovastatin with dextroamphetamine sulfate during the first trimester of pregnancy.
Labor and Delivery
No studies have been conducted in pregnant women on the effect of CADUET, amlodipine or atorvastatin on the mother or the fetus during labor or delivery, or on the duration of labor or delivery. Amlodipine has been shown to prolong the duration of labor in rats.
Nursing Mothers
It is not known whether the amlodipine component of CADUET is excreted in human milk. Nursing rat pups taking atorvastatin had plasma and liver drug levels of 50% and 40%, respectively, of that in their mother's milk. Because of the potential for adverse reactions in nursing infants, women taking CADUET should not breast-feed (see CONTRAINDICATIONS ).
Pediatric Use
There have been no studies conducted to determine the safety or effectiveness of CADUET in pediatric populations.
Studies with amlodipine: The effect of amlodipine on blood pressure in patients less than 6 years of age is not known.
Studies with atorvastatin: Safety and effectiveness in patients 10-17 years of age with heterozygous familial hypercholesterolemia have been evaluated in controlled clinical trials of 6 months duration in adolescent boys and postmenarchal girls. Patients treated with atorvastatin had an adverse experience profile generally similar to that of patients treated with placebo, the most common adverse experiences observed in both groups, regardless of causality assessment, were infections. Doses greater than 20 mg have not been studied in this patient population. In this limited controlled study, there was no detectable effect on growth or sexual maturation in boys or on menstrual cycle length in girls. See CLINICAL PHARMACOLOGY , Clinical Studies section; ADVERSE REACTIONS , Pediatric Patients ; and DOSAGE AND ADMINISTRATION , Pediatric Patients (10-17 years of age) with Heterozygous Familial Hypercholesterolemia . Adolescent females should be counseled on appropriate contraceptive methods while on atorvastatin therapy (see CONTRAINDICATIONS and PRECAUTIONS , Pregnancy ). Atorvastatin has not been studied in controlled clinical trials involving pre-pubertal patients or patients younger than 10 years of age.
Clinical efficacy with doses of atorvastatin up to 80 mg/day for 1 year have been evaluated in an uncontrolled study of patients with homozygous FH including 8 pediatric patients. See CLINICAL PHARMACOLOGY , Clinical Studies , Atorvastatin Effects in Homozygous Familial Hypercholesterolemia .
Geriatric Use
There have been no studies conducted to determine the safety or effectiveness of CADUET in geriatric populations.
In studies with amlodipine: Clinical studies of amlodipine did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection of the amlodipine component of CADUET for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy. Elderly patients have decreased clearance of amlodipine with a resulting increase of AUC of approximately 40-60%, and a lower initial dose may be required (see DOSAGE AND ADMINISTRATION ).
In studies with atorvastatin: The safety and efficacy of atorvastatin (10-80 mg) in the geriatric population (>/=65 years of age) was evaluated in the ACCESS study. In this 54-week open-label trial 1,958 patients initiated therapy with atorvastatin calcium 10 mg. Of these, 835 were elderly (>/=65 years) and 1,123 were non-elderly. The mean change in LDL-C from baseline after 6 weeks of treatment with atorvastatin calcium 10 mg was -38.2% in the elderly patients versus -34.6% in the non-elderly group.
The rates of discontinuation in patients on atorvastatin due to adverse events were similar between the two age groups. There were no differences in clinically relevant laboratory abnormalities between the age groups.
ADVERSE REACTIONS
CADUET
CADUET (amlodipine besylate/atorvastatin calcium) has been evaluated for safety in 1092 patients in double-blind placebo controlled studies treated for co-morbid hypertension and dyslipidemia. In general, treatment with CADUET was well tolerated. For the most part, adverse experiences have been mild or moderate in severity. In clinical trials with CADUET, no adverse experiences peculiar to this combination have been observed. Adverse experiences are similar in terms of nature, severity, and frequency to those reported previously with amlodipine and atorvastatin.
The following information is based on the clinical experience with amlodipine and atorvastatin.
The Amlodipine Component of CADUET
Amlodipine has been evaluated for safety in more than 11,000 patients in U.S. and foreign clinical trials. In general, treatment with amlodipine was well tolerated at doses up to 10 mg daily. Most adverse reactions reported during therapy with amlodipine were of mild or moderate severity. In controlled clinical trials directly comparing amlodipine (N=1730) in doses up to 10 mg to placebo (N=1250), discontinuation of amlodipine due to adverse reactions was required in only about 1.5% of patients and was not significantly different from placebo (about 1%). The most common side effects are headache and edema. The incidence (%) of side effects which occurred in a dose related manner are as follows:
Adverse Event amlodipine 2.5 mg
N=2755.0 mg
N=29610.0 mg
N=268Placebo
N=520Edema1.8 3.0 10.8 0.6 Dizziness1.1 3.4 3.4 1.5 Flushing0.7 1.4 2.6 0.0 Palpitation0.7 1.4 4.5 0.6
Other adverse experiences which were not clearly dose related but which were reported with an incidence greater than 1.0% in placebo-controlled clinical trials include the following:
Placebo-Controlled Studies
Adverse Eventamlodipine (%)
(N=1730)Placebo (%)
(N=1250)Headache7.3 7.8 Fatigue4.5 2.8 Nausea2.9 1.9 Abdominal Pain1.6 0.3 Somnolence1.4 0.6
For several adverse experiences that appear to be drug and dose related, there was a greater incidence in women than men associated with amlodipine treatment as shown in the following table:
Adverse Event amlodipine Placebo M=%
(N=1218)F=%
(N=512)M=%
(N=914)F=%
(N=336)Edema5.6 14.6 1.4 5.1 Flushing1.5 4.5 0.3 0.9 Palpitations1.4 3.3 0.9 0.9 Somnolence1.3 1.6 0.8 0.3
The following events occurred in </=1% but >0.1% of patients treated with amlodipine in controlled clinical trials or under conditions of open trials or marketing experience where a causal relationship is uncertain; they are listed to alert the physician to a possible relationship:
Cardiovascular: arrhythmia (including ventricular tachycardia and atrial fibrillation), bradycardia, chest pain, hypotension, peripheral ischemia, syncope, tachycardia, postural dizziness, postural hypotension, vasculitis.
Central and Peripheral Nervous System: hypoesthesia, neuropathy peripheral, paresthesia, tremor, vertigo.
Gastrointestinal: anorexia, constipation, dyspepsia, ** dysphagia, diarrhea, flatulence, pancreatitis, vomiting, gingival hyperplasia.
General: allergic reaction, asthenia, ** back pain, hot flushes, malaise, pain, rigors, weight gain, weight decrease.
Musculoskeletal System: arthralgia, arthrosis, muscle cramps, ** myalgia.
Psychiatric: sexual dysfunction (male ** and female), insomnia, nervousness, depression, abnormal dreams, anxiety, depersonalization.
Respiratory System: dyspnea, ** epistaxis.
Skin and Appendages: angioedema, erythema multi-forme, pruritus, ** rash, ** rash erythematous, rash maculopapular.
**These events occurred in less than 1% in placebo-controlled trials, but the incidence of these side effects was between 1% and 2% in all multiple dose studies.
Special Senses: abnormal vision, conjunctivitis, diplopia, eye pain, tinnitus.
Urinary System: micturition frequency, micturition disorder, nocturia.
Autonomic Nervous System: dry mouth, sweating increased.
Metabolic and Nutritional: hyperglycemia, thirst.
Hemopoietic: leukopenia, purpura, thrombocytopenia.
The following events occurred in </=0.1% of patients treated with amlodipine in controlled clinical trials or under conditions of open trials or marketing experience: cardiac failure, pulse irregularity, extrasystoles, skin discoloration, urticaria, skin dryness, alopecia, dermatitis, muscle weakness, twitching, ataxia, hypertonia, migraine, cold and clammy skin, apathy, agitation, amnesia, gastritis, increased appetite, loose stools, coughing, rhinitis, dysuria, polyuria, parosmia, taste perversion, abnormal visual accommodation, and xerophthalmia.
Other reactions occurred sporadically and cannot be distinguished from medications or concurrent disease states such as myocardial infarction and angina.
Amlodipine therapy has not been associated with clinically significant changes in routine laboratory tests. No clinically relevant changes were noted in serum potassium, serum glucose, total triglycerides, total cholesterol, HDL cholesterol, uric acid, blood urea nitrogen, or creatinine.
The following postmarketing event has been reported infrequently with amlodipine treatment where a causal relationship is uncertain: gynecomastia. In postmarketing experience, jaundice and hepatic enzyme elevations (mostly consistent with cholestasis or hepatitis) in some cases severe enough to require hospitalization have been reported in association with use of amlodipine.
Amlodipine has been used safely in patients with chronic obstructive pulmonary disease, well-compensated congestive heart failure, peripheral vascular disease, diabetes mellitus, and abnormal lipid profiles.
The Atorvastatin Component of CADUET
Atorvastatin is generally well-tolerated. Adverse reactions have usually been mild and transient. In controlled clinical studies of 2502 patients, <2% of patients were discontinued due to adverse experiences attributable to atorvastatin calcium. The most frequent adverse events thought to be related to atorvastatin calcium were constipation, flatulence, dyspepsia, and abdominal pain.
Clinical Adverse Experiences
Adverse experiences reported in >/=2% of patients in placebo-controlled clinical studies of atorvastatin, regardless of causality assessment, are shown in Table 10.
Table 10. Adverse Events in Placebo-Controlled Studies
(% of Patients)Body System/
Adverse EventPlacebo
N=270atorvastatin 10 mg
N=86320 mg
N=3640 mg
N=7980 mg
N=94BODY AS A WHOLEInfection10.0 10.3 2.8 10.1 7.4 Headache7.0 5.4 16.7 2.5 6.4 Accidental Injury3.7 4.2 0.0 1.3 3.2 Flu Syndrome1.9 2.2 0.0 2.5 3.2 Abdominal Pain0.7 2.8 0.0 3.8 2.1 Back Pain3.0 2.8 0.0 3.8 1.1 Allergic Reaction2.6 0.9 2.8 1.3 0.0 Asthenia1.9 2.2 0.0 3.8 0.0 DIGESTIVE SYSTEMConstipation1.8 2.1 0.0 2.5 1.1 Diarrhea1.5 2.7 0.0 3.8 5.3 Dyspepsia4.1 2.3 2.8 1.3 2.1 Flatulence3.3 2.1 2.8 1.3 1.1 RESPIRATORY SYSTEMSinusitis2.6 2.8 0.0 2.5 6.4 Pharyngitis1.5 2.5 0.0 1.3 2.1 SKIN AND APPENDAGESRash0.7 3.9 2.8 3.8 1.1 MUSCULOSKELETAL SYSTEMArthralgia1.5 2.0 0.0 5.1 0.0 Myalgia1.1 3.2 5.6 1.3 0.0 Anglo-Scandinavian Cardiac Outcomes Trial (ASCOT)
In ASCOT (see CLINICAL PHARMACOLOGY , Clinical Studies , Clinical Studies with Atorvastatin ) involving 10,305 participants treated with atorvastatin 10 mg daily (n=5,168) or placebo (n=5,137), the safety and tolerability profile of the group treated with atorvastatin was comparable to that of the group treated with placebo during a median of 3.3 years of follow-up.
The following adverse events were reported, regardless of causality assessment, in patients treated with atorvastatin in clinical trials. The events in italics occurred in >/=2% of patients and the events in plain type occurred in <2% of patients.
Body as a Whole: Chest pain , face edema, fever, neck rigidity, malaise, photosensitivity reaction, generalized edema.
Digestive System: Nausea , gastroenteritis, liver function tests abnormal, colitis, vomiting, gastritis, dry mouth, rectal hemorrhage, esophagitis, eructation, glossitis, mouth ulceration, anorexia, increased appetite, stomatitis, biliary pain, cheilitis, duodenal ulcer, dysphagia, enteritis, melena, gum hemorrhage, stomach ulcer, tenesmus, ulcerative stomatitis, hepatitis, pancreatitis, cholestatic jaundice.
Respiratory System: Bronchitis, rhinitis , pneumonia, dyspnea, asthma, epistaxis.
Nervous System: Insomnia, dizziness , paresthesia, somnolence, amnesia, abnormal dreams, libido decreased, emotional lability, incoordination, peripheral neuropathy, torticollis, facial paralysis, hyperkinesia, depression, hypesthesia, hypertonia.
Musculoskeletal System: Arthritis , leg cramps, bursitis, tenosynovitis, myasthenia, tendinous contracture, myositis.
Skin and Appendages: Pruritus, contact dermatitis, alopecia, dry skin, sweating, acne, urticaria, eczema, seborrhea, skin ulcer.
Urogenital System: Urinary tract infection , urinary frequency, cystitis, hematuria, impotence, dysuria, kidney calculus, nocturia, epididymitis, fibrocystic breast, vaginal hemorrhage, albuminuria, breast enlargement, metrorrhagia, nephritis, urinary incontinence, urinary retention, urinary urgency, abnormal ejaculation, uterine hemorrhage.
Special Senses: Amblyopia, tinnitus, dry eyes, refraction disorder, eye hemorrhage, deafness, glaucoma, parosmia, taste loss, taste perversion.
Cardiovascular System: Palpitation, vasodilatation, syncope, migraine, postural hypotension, phlebitis, arrhythmia, angina pectoris, hypertension.
Metabolic and Nutritional Disorders: Peripheral edema , hyperglycemia, creatine phosphokinase increased, gout, weight gain, hypoglycemia.
Hemic and Lymphatic System: Ecchymosis, anemia, lymphadenopathy, thrombocytopenia, petechia.
Postintroduction Reports with Atorvastatin
Adverse events associated with atorvastatin therapy reported since market introduction, that are not listed above, regardless of causality assessment, include the following: anaphylaxis, angioneurotic edema, bullous rashes (including erythema multiforme, Stevens-Johnson syndrome, and toxic epidermal necrolysis), and rhabdomyolysis.
Pediatric Patients (ages 10-17 years)
In a 26-week controlled study in boys and postmenarchal girls (n=140), the safety and tolerability profile of atorvastatin 10 to 20 mg daily was generally similar to that of placebo (see CLINICAL PHARMACOLOGY , Clinical Studies section and PRECAUTIONS , Pediatric Use ).
OVERDOSAGE
There is no information on overdosage with CADUET in humans.
Information on Amlodipine
Single oral doses of amlodipine maleate equivalent to 40 mg amlodipine/kg and 100 mg amlodipine/kg in mice and rats, respectively, caused deaths. Single oral amlodipine maleate doses equivalent to 4 or more mg amlodipine/kg in dogs (11 or more times the maximum recommended clinical dose on a mg/m 2 basis) caused a marked peripheral vasodilation and hypotension.
Overdosage might be expected to cause excessive peripheral vasodilation with marked hypotension and possibly a reflex tachycardia. In humans, experience with intentional overdosage of amlodipine is limited. Reports of intentional overdosage include a patient who ingested 250 mg and was asymptomatic and was not hospitalized; another (120 mg) was hospitalized, underwent gastric lavage and remained normotensive; the third (105 mg) was hospitalized and had hypotension (90/50 mmHg) which normalized following plasma expansion. A patient who took 70 mg amlodipine and an unknown quantity of benzodiazepine in a suicide attempt developed shock which was refractory to treatment and died the following day with abnormally high benzodiazepine plasma concentration. A case of accidental drug overdose has been documented in a 19-month-old male who ingested 30 mg amlodipine (about 2 mg/kg). During the emergency room presentation, vital signs were stable with no evidence of hypotension, but a heart rate of 180 bpm. Ipecac was administered 3.5 hours after ingestion and on subsequent observation (overnight) no sequelae were noted.
If massive overdose should occur, active cardiac and respiratory monitoring should be instituted. Frequent blood pressure measurements are essential. Should hypotension occur, cardiovascular support including elevation of the extremities and the judicious administration of fluids should be initiated. If hypotension remains unresponsive to these conservative measures, administration of vasopressors (such as phenylephrine) should be considered with attention to circulating volume and urine output. Intravenous calcium gluconate may help to reverse the effects of calcium entry blockade. As amlodipine is highly protein bound, hemodialysis is not likely to be of benefit.
Information on Atorvastatin
There is no specific treatment for atorvastatin overdosage. In the event of an overdose, the patient should be treated symptomatically, and supportive measures instituted as required. Due to extensive drug binding to plasma proteins, hemodialysis is not expected to significantly enhance atorvastatin clearance.
DOSAGE AND ADMINISTRATION
Dosage of CADUET must be individualized on the basis of both effectiveness and tolerance for each individual component in the treatment of hypertension/angina and hyperlipidemia.
Amlodipine (Hypertension or angina)
Adults: The usual initial antihypertensive oral dose of amlodipine is 5 mg once daily with a maximum dose of 10 mg once daily. Small, fragile, or elderly individuals, or patients with hepatic insufficiency may be started on 2.5 mg once daily and this dose may be used when adding amlodipine to other antihypertensive therapy.
Dosage should be adjusted according to each patient's need. In general, titration should proceed over 7 to 14 days so that the physician can fully assess the patient's response to each dose level. Titration may proceed more rapidly, however, if clinically warranted, provided the patient is assessed frequently.
The recommended dose of amlodipine for chronic stable or vasospastic angina is 5-10 mg, with the lower dose suggested in the elderly and in patients with hepatic insufficiency. Most patients will require 10 mg for adequate effect. See ADVERSE REACTIONS section for information related to dosage and side effects.
Children: The effective antihypertensive oral dose of amlodipine in pediatric patients ages 6-17 years is 2.5 mg to 5 mg once daily. Doses in excess of 5 mg daily have not been studied in pediatric patients. See CLINICAL PHARMACOLOGY .
Atorvastatin (Hyperlipidemia)
The patient should be placed on a standard cholesterol-lowering diet before receiving atorvastatin and should continue on this diet during treatment with atorvastatin.
Hypercholesterolemia (Heterozygous Familial and Nonfamilial) and Mixed Dyslipidemia (Fredrickson Types IIa and IIb )
The recommended starting dose of atorvastatin is 10 or 20 mg once daily. Patients who require a large reduction in LDL-C (more than 45%) may be started at 40 mg once daily. The dosage range of atorvastatin is 10 to 80 mg once daily. Atorvastatin can be administered as a single dose at any time of the day, with or without food. The starting dose and maintenance doses of atorvastatin should be individualized according to patient characteristics such as goal of therapy and response (see NCEP Guidelines , summarized in Table 8). After initiation and/or upon titration of atorvastatin, lipid levels should be analyzed within 2 to 4 weeks and dosage adjusted accordingly.
Since the goal of treatment is to lower LDL-C, the NCEP recommends that LDL-C levels be used to initiate and assess treatment response. Only if LDL-C levels are not available, should total-C be used to monitor therapy.
Heterozygous Familial Hypercholesterolemia in Pediatric Patients (10-17 years of age)
The recommended starting dose of atorvastatin is 10 mg/day; the maximum recommended dose is 20 mg/day (doses greater than 20 mg have not been studied in this patient population). Doses should be individualized according to the recommended goal of therapy (see NCEP Pediatric Panel Guidelines 1 , CLINICAL PHARMACOLOGY , and INDICATIONS AND USAGE ). Adjustments should be made at intervals of 4 weeks or more.
Homozygous Familial Hypercholesterolemia
The dosage of atorvastatin in patients with homozygous FH is 10 to 80 mg daily. Atorvastatin should be used as an adjunct to other lipid-lowering treatments (e.g., LDL apheresis) in these patients or if such treatments are unavailable. Note: a 2.5/80 mg CADUET tablet is not available. Management of patients needing a 2.5/80 mg combination requires individual assessments of dyslipidemia and therapy with the individual components as a 2.5/80 mg CADUET tablet is not available.
1 National Cholesterol Education Program (NCEP): Highlights of the Report of the Expert Panel on Blood Cholesterol Levels in Children Adolescents. Pediatrics . 89(3):495-501. 1992.
Concomitant Therapy
Atorvastatin may be used in combination with a bile acid binding resin for additive effect. The combination of HMG-CoA reductase inhibitors and fibrates should generally be avoided (see WARNINGS , Skeletal Muscle , and PRE-CAUTIONS , Drug Interactions for other drug-drug interactions).
Dosage in Patients With Renal Insufficiency
Renal disease does not affect the plasma concentrations nor LDL-C reduction of atorvastatin; thus, dosage adjustment in patients with renal dysfunction is not necessary (see CLINICAL PHARMACOLOGY , Pharmacokinetics ).
CADUET
CADUET may be substituted for its individually titrated components. Patients may be given the equivalent dose of CADUET or a dose of CADUET with increased amounts of amlodipine, atorvastatin or both for additional antianginal effects, blood pressure lowering, or lipid lowering effect.
CADUET may be used to provide additional therapy for patients already on one of its components. As initial therapy for one indication and continuation of treatment of the other, the recommended starting dose of CADUET should be selected based on the continuation of the component being used and the recommended starting dose for the added monotherapy.
CADUET may be used to initiate treatment in patients with hyperlipidemia and either hypertension or angina. The recommended starting dose of CADUET should be based on the appropriate combination of recommendations for the monotherapies. The maximum dose of the amlodipine component of CADUET is 10 mg once daily. The maximum dose of the atorvastatin component of CADUET is 80 mg once daily.
See above for detailed information related to the dosing and administration of amlodipine and atorvastatin.
HOW SUPPLIED
CADUET ® tablets contain amlodipine besylate and atorvastatin calcium equivalent to amlodipine and atorvastatin in the dose strengths described below.
CADUET tablets are differentiated by tablet color/size and are engraved with "Pfizer" on one side and a unique number on the other side. CADUET tablets are supplied for oral administration in the following strengths and package configurations:
Table 11. CADUET Packaging Configurations CADUET Package
ConfigurationTablet Strength
(amlodipine besylate/
atorvastatin calcium) mgNDC #EngravingTablet ColorBottle of 30 2.5/10 0069-2960-30CDT 251WhiteBottle of 30 2.5/20 0069-2970-30CDT 252WhiteBottle of 30 2.5/40 0069-2980-30CDT 254WhiteBottle of 30 5/10 0069-2150-30CDT 051WhiteBottle of 30 5/20 0069-2170-30CDT 052WhiteBottle of 30 5/40 0069-2190-30CDT 054WhiteBottle of 30 5/80 0069-2260-30CDT 058WhiteBottle of 30 10/10 0069-2160-30CDT 101BlueBottle of 30 10/20 0069-2180-30CDT 102BlueBottle of 30 10/40 0069-2250-30 CDT 104 BlueBottle of 30 10/80 0069-2270-30 CDT 108 Blue
Store at 25°C (77°F); excursions permitted to 15-30°C (59-86°F) [see USP Controlled Room Temperature].
Rx only © 2004 Pfizer Ireland Pharmaceuticals
Manufactured by:
Pfizer Ireland Pharmaceuticals
Dublin, Ireland
Distributed by
Pfizer Labs
Division of Pfizer Inc, NY, NY 10017
LAB-0276-3.0 Revised October 2004
Subscribe to the "News" RSS Feed 
TOP ۞