-
Lamisil Tablets (Novartis)
Prescribing Information
The following prescribing information is based on official labeling in effect August 2005.
DESCRIPTION
LAMISIL® (terbinafine hydrochloride tablets) Tablets contain the synthetic allylamine antifungal compoundterbinafine hydrochloride.
Chemically, terbinafine hydrochloride is (E)- N -(6,6-dimethyl-2-hepten-4-ynyl)- N -methyl-1-naphthalenemeth-anamine hydrochloride. The empirical formula C 21 H 26 ClN with a molecular weight of 327.90, and the following structural formula:
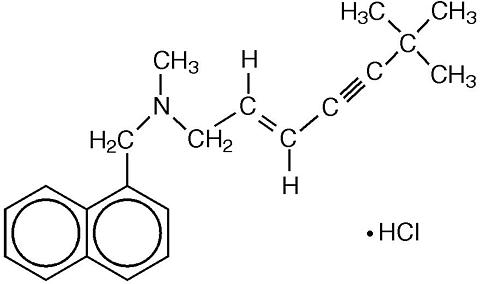
Terbinafine hydrochloride is a white to off-white fine crystalline powder. It is freely soluble in methanol and methylene chloride, soluble in ethanol, and slightly soluble in water.
Each tablet contains:
Active Ingredients: terbinafine hydrochloride (equivalent to 250 mg base)
Inactive Ingredients: colloidal silicon dioxide, NF; hydroxypropyl methylcellulose, USP; magnesium stearate, NF; microcrystalline cellulose, NF; sodium starch glycolate, NF
CLINICAL PHARMACOLOGY
Pharmacokinetics
Following oral administration, terbinafine is well absorbed (>70%) and the bioavailability of LAMISIL® (terbinafine hydrochloride tablets) Tablets as a result of first-pass metabolism is approximately 40%. Peak plasma concentrations of 1 µg/mL appear within 2 hours after a single 250 mg dose; the AUC (area under the curve) is approximately 4.56 µg·h/mL. An increase in the AUC of terbinafine of less than 20% is observed when LAMISIL® is administered with food. No clinically relevant age-dependent changes in steady-state plasma concentrations of terbinafine have been reported. In patients with renal impairment (creatinine clearance </=50 mL/min) or hepatic cirrhosis, the clearance ofterbinafine is decreased by approximately 50% compared to normal volunteers. No effect of gender on the blood levels of terbinafine was detected in clinical trials. In plasma,terbinafine is >99% bound to plasma proteins and there are no specific binding sites. At steady-state, in comparison to a single dose, the peak concentration of terbinafine is 25% higher and plasma AUC increases by a factor of 2.5; the increase in plasma AUC is consistent with an effective half-life of ~36 hours. Terbinafine is distributed to the sebum and skin. A terminal half-life of 200-400 hours may represent the slow elimination of terbinafine from tissues such as skin and adipose. Prior to excretion, terbinafine is extensively metabolized. No metabolites have been identified that have antifungal activity similar to terbinafine. Approximately 70% of the administered dose is eliminated in the urine.
Microbiology
Terbinafine hydrochloride is a synthetic allylamine derivative. Terbinafine hydrochloride is hypothesized to act by inhibiting squalene epoxidase, thus blocking the biosynthesis of ergosterol, an essential component of fungal cell membranes. In vitro , mammalian squalene epoxidase is only inhibited at higher (4000 fold) concentrations than is needed for inhibition of the dermatophyte enzyme. Depending on the concentration of the drug and the fungal species test in vitro , terbinafine hydrochloride may be fungicidal. However, the clinical significance of in vitro data is unknown.
Terbinafine has been shown to be active against most strains of the following microorganisms both in vitro and in clinical infections as described in the INDICATIONS AND USAGE section:
Trichophyton mentagrophytes
Trichophyton rubrum
The following in vitro data are available, but their clinical significance is unknown. In vitro , terbinafine exhibits satisfactory MIC's against most strains of the following microorganisms; however, the safety and efficacy of terbinafine in treating clinical infections due to these microorganisms have not been established in adequate and well-controlled clinical trials:
Candida albicans
Epidermophyton floccosum
Scopulariopsis brevicaulis
CLINICAL STUDIES
The efficacy of LAMISIL® (terbinafine hydrochloridetablets) Tablets in the treatment of onychomycosis is illustrated by the response of patients with toenail and/or fingernail infections who participated in three US/Canadian placebo-controlled clinical trials.
Results of the first toenail study, as assessed at week 48 (12 weeks of treatment with 36 weeks follow-up after completion of therapy), demonstrated mycological cure, defined as simultaneous occurrence of negative KOH plus negative culture, in 70% of patients. Fifty-nine percent (59%) of patients experienced effective treatment (mycological cure plus 0% nail involvement or >5mm of new unaffected nail growth); 38% of patients demonstrated mycological cure plus clinical cure (0% nail involvement).
In a second toenail study of dermatophytic onychomycosis, in which non-dermatophytes were also cultured, similar efficacy against the dermatophytes was demonstrated. The pathogenic role of the non-dermatophytes cultured in the presence of dermatophytic onychomycosis has not been established. The clinical significance of this association is unknown.
Results of the fingernail study, as assessed at week 24 (6 weeks of treatment with 18 weeks follow-up after completion of therapy), demonstrated mycological cure in 79% of patients, effective treatment in 75% of the patients, and mycological cure plus clinical cure in 59% of the patients.
The mean time to overall success was approximately 10 months for the first toenail study and 4 months for the fingernail study. In the first toenail study, for patients evaluated at least six months after achieving clinical cure and at least one year after completing LAMISIL® therapy, the clinical relapse rate was approximately 15%.
INDICATIONS AND USAGE
LAMISIL® (terbinafine hydrochloride tablets) Tablets are indicated for the treatment of onychomycosis of the toenail or fingernail due to dermatophytes (tinea unguium) (see DOSAGE AND ADMINISTRATION and CLINICAL STUDIES ).
Prior to initiating treatment, appropriate nail specimens for laboratory testing (KOH preparation, fungal culture, or nail biopsy) should be obtained to confirm the diagnosis of onychomycosis.
CONTRAINDICATIONS
LAMISIL® (terbinafine hydrochloride tablets) Tablets are contraindicated in individuals with hypersensitivity toterbinafine or to any other ingredients of the formulation.
WARNINGS
Rare cases of liver failure, some leading to death or liver transplant, have occurred with the use of LAMISIL® (terbinafine hydrochloride tablets) Tablets for the treatment of onychomycosis in individuals with and without pre-existing liver disease.
In the majority of liver cases reported in association with LAMISIL® use, the patients had serious underlying systemic conditions and an uncertain causal association with LAMISIL®. The severity of hepatic events and/or their outcome may be worse in patients with active or chronic liver disease (see PRECAUTIONS ). Treatment with LAMISIL® Tablets should be discontinued if biochemical or clinical evidence of liver injury develops (see PRECAUTIONS below). There have been isolated reports of serious skin reactions (e.g., Stevens-Johnson Syndrome and toxic epidermal necrolysis). If progressive skin rash occurs, treatment with LAMISIL® should be discontinued.
PRECAUTIONS
General
LAMISIL® (terbinafine hydrochloride tablets) Tablets are not recommended for patients with chronic or active liver disease. Before prescribing LAMISIL® Tablets, pre-existing liver disease should be assessed. Hepatotoxicity may occur in patients with and without pre-existing liver disease. Pretreatment serum transaminase (ALT and AST) tests are advised for all patients before taking LAMISIL® Tablets. Patients prescribed LAMISIL® Tablets should be warned to report immediately to their physician any symptoms of persistent nausea, anorexia, fatigue, vomiting, right upper abdominal pain or jaundice, dark urine or pale stools (see WARNINGS ). Patients with these symptoms should discontinue taking oral terbinafine, and the patient's liver function should be immediately evaluated.
In patients with renal impairment (creatinine clearance </=50 mL/min), the use of LAMISIL® has not been adequately studied, and therefore, is not recommended (see CLINICAL PHARMACOLOGY , Pharmacokinetics ).
During post-marketing experience, precipitation and exacerbation of cutaneous and systemic lupus erythematosus have been reported infrequently in patients taking LAMISIL®. LAMISIL® therapy should be discontinued in patients with clinical signs and symptoms suggestive of lupus erythematosus.
Changes in the ocular lens and retina have been reported following the use of LAMISIL® (terbinafine hydrochloride tablets) Tablets in controlled trials. The clinical significance of these changes is unknown.
Transient decreases in absolute lymphocyte counts (ALC) have been observed in controlled clinical trials. In placebo-controlled trials, 8/465 LAMISIL®-treated patients (1.7%) and 3/137 placebo-treated patients (2.2%) had decreases in ALC to below 1000/mm 3 on two or more occasions. The clinical significance of this observation is unknown. However, in patients with known or suspected immunodeficiency, physicians should consider monitoring complete blood counts in individuals using LAMISIL® therapy for greater than six weeks.
Isolated cases of severe neutropenia have been reported. These were reversible upon discontinuation of LAMISIL®, with or without supportive therapy. If clinical signs and symptoms suggestive of secondary infection occur, a complete blood count should be obtained. If the neutrophil count is </=1,000 cells/mm 3 , LAMISIL® should be discontinued and supportive management started.
Drug Interactions
In vivo studies have shown that terbinafine is an inhibitor of the CYP450 2D6 isozyme. Coadministration of LAMISIL® with drugs predominantly metabolized by the CYP450 2D6 isozyme (e.g., certain members of the following drug classes, tricyclic antidepressants, selective serotonin reuptake inhibitors, beta-blockers, antiarrhythmics class 1C and monoamine oxidase inhibitors Type B) should be done with careful monitoring and may require a reduction in dose of the 2D6-metabolized drug. In a study to assess the effects of terbinafine on desipramine in healthy volunteers characterized as normal metabolizers, the administration of terbinafine resulted in a 2-fold increase in C max and a 5-fold increase in AUC. In this study, these effects were shown to persist at the last observation at 4 weeks after discontinuation of LAMISIL®.
In vitro studies with human liver microsomes showed that terbinafine does not inhibit the metabolism of tolbutamide, ethinylestradiol, ethoxycoumarin, and cyclosporine.
In vivo drug-drug interaction studies conducted in healthy volunteer subjects showed that terbinafine does not affect the clearance of antipyrine or digoxin. Terbinafine decreases the clearance of caffeine by 19%. Terbinafine increases the clearance of cyclosporine by 15%.
There have been spontaneous reports of increase or decrease in prothrombin times in patients concomitantly taking oral terbinafine and warfarin, however, a causal relationship between LAMISIL® Tablets and these changes has not been established.
Terbinafine clearance is increased 100% by rifampin, a CYP450 enzyme inducer, and decreased 33% by cimetidine, a CYP450 enzyme inhibitor. Terbinafine clearance is unaffected by cyclosporine.
There is no information available from adequate drug-drug interaction studies with the following classes of drugs: oral contraceptives, hormone replacement therapies, hypoglycemics, theophyllines, phenytoins, thiazide diuretics, and calcium channel blockers.
Carcinogenesis, Mutagenesis, Impairment of Fertility
In a 28-month oral carcinogenicity study in rats, an increase in the incidence of liver tumors was observed in males at the highest dose tested, 69 mg/kg/day [2 × the Maximum Recommended Human Dose (MRHD) based on AUC comparisons of the parent terbinafine]; however, even though dose-limiting toxicity was not achieved at the highest tested dose, higher doses were not tested.
The results of a variety of in vitro (mutations in E. coli and S. typhimurium , DNA repair in rat hepatocytes, mutagenicity in Chinese hamster fibroblasts, chromosome aberration and sister chromatid exchanges in Chinese hamster lung cells), and in vivo (chromosome aberration in Chinese hamsters, micronucleus test in mice) genotoxicity tests gave no evidence of a mutagenic or clastogenic potential. Oral reproduction studies in rats at doses up to 300 mg/kg/day (approximately 12 × the MRHD based on body surface area comparisons, BSA) did not reveal any specific effects on fertility or other reproductive parameters. Intravaginal application of terbinafine hydrochloride at 150 mg/day in pregnant rabbits did not increase the incidence of abortions or premature deliveries nor affect fetal parameters.
Pregnancy
Pregnancy Category B: Oral reproduction studies have been performed in rabbits and rats at doses up to300 mg/kg/day (12 × to 23 × the MRHD, in rabbits and rats, respectively, based on BSA) and have revealed no evidence of impaired fertility or harm to the fetus due to terbinafine. There are, however, no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, and because treatment of onychomycosis can be postponed until after pregnancy is completed, it is recommended that LAMISIL® not be initiated during pregnancy.
Nursing Mothers
After oral administration, terbinafine is present in breast milk of nursing mothers. The ratio of terbinafine in milk to plasma is 7:1. Treatment with LAMISIL® is not recommended in nursing mothers.
Pediatric Use
The safety and efficacy of LAMISIL® have not been established in pediatric patients.
ADVERSE REACTIONS
The most frequently reported adverse events observed in the three US/Canadian placebo-controlled trials are listed in the table below. The adverse events reported encompass gastrointestinal symptoms (including diarrhea, dyspepsia, and abdominal pain), liver test abnormalities, rashes, urticaria, pruritus, and taste disturbances. In general, the adverse events were mild, transient, and did not lead to discontinuation from study participation.
Adverse Event Discontinuation LAMISIL ®
(%)
n=465Placebo
(%)
n=137LAMISIL ®
(%)
n=465Placebo
(%)
n=137Headache12.9 9.5 0.2 0.0 Gastrointestinal Symptoms:Diarrhea5.6 2.9 0.6 0.0 Dyspepsia4.3 2.9 0.4 0.0 Abdominal Pain2.4 1.5 0.4 0.0 Nausea2.6 2.9 0.2 0.0 Flatulence2.2 2.2 0.0 0.0 Dermatological Symptoms:Rash5.6 2.2 0.9 0.7 Pruritus2.8 1.5 0.2 0.0 Urticaria1.1 0.0 0.0 0.0 Liver EnzymeAbnormalities *3.3 1.4 0.2 0.0 Taste Disturbance2.8 0.7 0.2 0.0 Visual Disturbance1.1 1.5 0.9 0.0 *Liver enzyme abnormalities >/=2 × the upper limit of the normal range.Rare adverse events, based on worldwide experience with LAMISIL® (terbinafine hydrochloride tablets) Tablets use, include: idiosyncratic and symptomatic hepatic injury and more rarely, cases of liver failure, some leading to death or liver transplant, (see WARNINGS and PRECAUTIONS ), serious skin reactions (see WARNINGS ), severe neutropenia (see PRECAUTIONS ), thrombocytopenia, angioedema and allergic reactions (including anaphylaxis). Psoriasiform eruptions or exacerbation of psoriasis, acute generalized exanthematous pustulosis and precipitation and exacerbation of cutaneous and systemic lupus erythematosus have been reported infrequently in patients taking LAMISIL®. Uncommonly, LAMISIL® may cause taste disturbance (including taste loss) which usually recovers within several weeks after discontinuation of the drug. There have been isolated reports of prolonged (greater than one year) taste disturbance. Rarely, taste disturbances associated with oral terbinafine have been reported to be severe enough to result in decreased food intake leading to significant and unwanted weight loss.
Other adverse reactions which have been reported include malaise, fatigue, vomiting, arthralgia, myalgia, and hair loss.
Clinical adverse effects reported spontaneously since the drug was marketed include altered prothrombin time (prolongation and reduction) in patients concomitantly treated with warfarin and LAMISIL® Tablets and agranulocytosis (rare).
OVERDOSAGE
Clinical experience regarding overdose with LAMISIL® (terbinafine hydrochloride tablets) Tablets is limited. Doses up to 5 grams (20 times the therapeutic daily dose) have been taken without inducing serious adverse reactions. The symptoms of overdose included nausea, vomiting, abdominal pain, dizziness, rash, frequent urination, and headache.
DOSAGE AND ADMINISTRATION
LAMISIL® (terbinafine hydrochloride tablets) Tablets, one 250 mg tablet, should be taken once daily for 6 weeks by patients with fingernail onychomycosis. LAMISIL®, one 250 mg tablet, should be taken once daily for 12 weeks by patients with toenail onychomycosis. The optimal clinical effect is seen some months after mycological cure and cessation of treatment. This is related to the period required for outgrowth of healthy nail.
HOW SUPPLIED
LAMISIL®
(terbinafine hydrochloride tablets)
Tablets
Supplied as white to yellow-tinged white circular, bi-convex, bevelled tablets containing 250 mg of terbinafine imprinted with "LAMISIL" in circular form on one side and code "250" on the other.
Bottles of 100 tablets NDC 0078-0179-05
Bottles of 30 tablets NDC 0078-0179-15
Store tablets below 25°C (77°F); in a tight container. Protect from light.
ANIMAL TOXICOLOGY
A wide range of in vivo studies in mice, rats, dogs, and monkeys, and in vitro studies using rat, monkey, and human hepatocytes suggest that peroxisome proliferation in the liver is a rat-specific finding. However, other effects, including increased liver weights and APTT, occurred in dogs and monkeys at doses giving Css trough levels of the parentterbinafine 2-3 × those seen in humans at the MRHD. Higher doses were not tested.
T2005-37
5000386
Distributed by:
Novartis Pharmaceuticals Corporation
East Hanover, New Jersey 07936
REV: AUGUST 2005
Subscribe to the "News" RSS Feed 
TOP ۞