-
Lupron Injection Pediatric (TAP)
DESCRIPTION
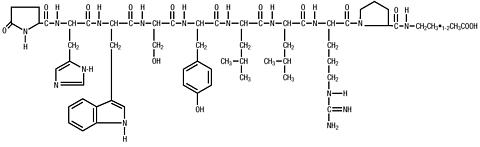
Leuprolide acetate is a synthetic nonapeptide analog of naturally occurring gonadotropin releasing hormone (GnRH or LH-RH). The analog possesses greater potency than the natural hormone. The chemical name is 5- oxo -L-prolyl-L-histidyl-L-tryptophyl-L-seryl-L-tyrosyl-D-leucyl-L-leucyl-L-arginyl-N-ethyl-L-prolinamide acetate (salt) with the following structural formula:
LUPRON INJECTION is a sterile, aqueous solution intended for daily subcutaneous injection. It is available in a 2.8 mL multiple dose vial containing leuprolide acetate (5 mg/mL), sodium chloride, USP (6.3 mg/mL) for tonicity adjustment, benzyl alcohol, NF as a preservative (9 mg/mL), and water for injection, USP. The pH may have been adjusted with sodium hydroxide, NF and/or acetic acid, NF.
CLINICAL PHARMACOLOGY
Leuprolide acetate, a GnRH agonist, acts as a potent inhibitor of gonadotropin secretion when given continuously and in therapeutic doses. Animal and human studies indicate that following an initial stimulation of gonadotropins, chronic administration of leuprolide acetate results in suppression of ovarian and testicular steroidogenesis. This effect is reversible upon discontinuation of drug therapy.
Leuprolide acetate is not active when given orally.
Pharmacokinetics
A pharmacokinetic study of leuprolide acetate in children has not been performed.
Absorption: In adults, bioavailability by subcutaneous administration is comparable to that by intravenous administration.
Distribution: The mean steady-state volume of distribution of leuprolide following intravenous bolus administration to healthy adult male volunteers was 27 L. In vitro binding to human plasma proteins ranged from 43% to 49%.
Metabolism: In healthy adult male volunteers, a 1 mg bolus of leuprolide administered intravenously revealed that the mean systemic clearance was 7.6 L/h, with a terminal elimination half-life of approximately 3 hours based on a two compartment model.
In rats and dogs, administration of 14 C-labeled leuprolide was shown to be metabolized to smaller inactive peptides, a pentapeptide (Metabolite I), tripeptides (Metabolites II and III) and a dipeptide (Metabolite IV). These fragments may be further catabolized.
The major metabolite (M-I) plasma concentrations measured in 5 prostate cancer patients reached maximum concentration 2 to 6 hours after dosing and were approximately 6% of the peak parent drug concentration. One week after dosing, mean plasma M-I concentrations were approximately 20% of mean leuprolide concentrations.
Excretion: Following administration of LUPRON DEPOT 3.75 mg to three adult patients, less than 5% of the dose was recovered as parent and M-I metabolite in the urine.
Special Populations: The pharmacokinetics of the drug in hepatically and renally impaired patients has not been determined.
Drug Interactions: No pharmacokinetic-based drug-drug interaction studies have been conducted with leuprolide acetate. However, because leuprolide acetate is a peptide that is primarily degraded by peptidase and the drug is only about 46% bound to plasma proteins, drug interactions would not be expected to occur.
CLINICAL STUDIES
In children with central precocious puberty (CPP), stimulated and basal gonadotropins are reduced to prepubertal levels. Testosterone and estradiol are reduced to prepubertal levels in males and females respectively. Reduction of gonadotropins will allow for normal physical and psychological growth and development. Natural maturation occurs when gonadotropins return to pubertal levels following discontinuation of leuprolide acetate.
The following physiologic effects have been noted with the chronic administration of leuprolide acetate in this patient population.
- Skeletal Growth. A measurable increase in body length can be noted since the epiphyseal plates will not close prematurely.
- Organ Growth. Reproductive organs will return to a prepubertal state.
- Menses. Menses, if present, will cease.
INDICATIONS AND USAGE
LUPRON INJECTION is indicated in the treatment of children with central precocious puberty. Children should be selected using the following criteria:
- Clinical diagnosis of CPP (idiopathic or neurogenic) with onset of secondary sexual characteristics earlier than 8 years in females and 9 years in males.
-
Clinical diagnosis should be confirmed prior to initiation of therapy:
- Confirmation of diagnosis by a pubertal response to a GnRH stimulation test. The sensitivity and methodology of this assay must be understood.
- Bone age advanced 1 year beyond the chronological age.
-
Baseline evaluation should also include:
- Height and weight measurements.
- Sex steroid levels.
- Adrenal steroid level to exclude congenital adrenal hyperplasia.
- Beta human chorionic gonadotropin level to rule out a chorionic gonadotropin secreting tumor.
- Pelvic/adrenal/testicular ultrasound to rule out a steroid secreting tumor.
- Computerized tomography of the head to rule out intracranial tumor.
CONTRAINDICATIONS
- Hypersensitivity to GnRH, GnRH agonist analogs or any of the excipients in LUPRON INJECTION. Reports of anaphylactic reactions to synthetic GnRH (Factrel) or GnRH agonist analogs have been reported in the medical literature 1 .
- LUPRON is contraindicated in women who are or may become pregnant while receiving the drug. LUPRON may cause fetal harm when administered to a pregnant woman. Major fetal abnormalities were observed in rabbits but not in rats after administration of leuprolide acetate throughout gestation. There was increased fetal mortality and decreased fetal weights in rats and rabbits. (See PRECAUTIONS , Pregnancy, Teratogenic Effects section.) The effects on fetal mortality are expected consequences of the alterations in hormonal levels brought about by this drug. Therefore, the possibility exists that spontaneous abortion may occur if the drug is administered during pregnancy. If this drug is administered during pregnancy or if the patient becomes pregnant while taking any formulation of LUPRON, the patient should be apprised of the potential hazard to the fetus.
WARNINGS
During the early phase of therapy, gonadotropins and sex steroids rise above baseline because of the natural stimulatory effect of the drug. Therefore, an increase in clinical signs and symptoms may be observed (see CLINICAL PHARMACOLOGY section).
Noncompliance with drug regimen or inadequate dosing may result in inadequate control of the pubertal process. The consequences of poor control include the return of pubertal signs such as menses, breast development, and testicular growth. The long-term consequences of inadequate control of gonadal steroid secretion are unknown, but may include a further compromise of adult stature.
PRECAUTIONS
Patients with known allergies to benzyl alcohol, an ingredient of the vehicle of LUPRON INJECTION, may present symptoms of hypersensitivity, usually local, in the form of erythema and induration at the injection site.
Information for Parents:
Prior to starting therapy with LUPRON INJECTION, the parent or guardian must be aware of the importance of continuous therapy. Adherence to daily drug administration schedules must be accepted if therapy is to be successful. Irregular dosing could restart the maturation process.
- During the first 2 months of therapy, a female may experience menses or spotting. If bleeding continues beyond the second month, notify the physician.
- Any irritation at the injection site should be reported to the physician immediately. If the child has experienced an allergic reaction to other drugs like LUPRON, this drug should not be used.
- Report any unusual signs or symptoms to the physician, like continued pubertal changes, substantial mood swings or behavioral changes.
Laboratory Tests: Response to leuprolide acetate should be monitored 1-2 months after the start of therapy with a GnRH stimulation test and sex steroid levels. Measurement of bone age for advancement should be done every 6-12 months.
Sex steroids may increase or rise above prepubertal levels if the dose is inadequate (see WARNINGS section). Once a therapeutic dose has been established, gonadotropin and sex steroid levels will decline to prepubertal levels.
Drug Interactions: See CLINICAL PHARMACOLOGY , Pharmacokinetics section.
Drug/Laboratory Test Interactions: Administration of leuprolide acetate in therapeutic doses results in suppression of the pituitary-gonadal system. Normal function is usually restored within 4 to 12 weeks after treatment is discontinued.
Carcinogenesis, Mutagenesis, Impairment of Fertility: A two-year carcinogenicity study was conducted in rats and mice. In rats, a dose-related increase of benign pituitary hyperplasia and benign pituitary adenomas was noted at 24 months when the drug was administered subcutaneously at high daily doses of 0.6 to 4 mg/kg (>100 times the clinical doses of 7.5 to 15 mg/month based on body surface area). There was a significant but not dose-related increase of pancreatic islet-cell adenomas in females and of testes interstitial cell adenomas in males (highest incidence in the low dose group). In mice, no leuprolide acetate-induced tumors or pituitary abnormalities were observed at daily dose as high as 60 mg/kg (>5000 times the clinical doses based on body surface area). Adult patients have been treated with leuprolide acetate for up to three years with doses as high as 10 mg/day and for two years with doses as high as 20 mg/day without demonstrable pituitary abnormalities.
Although no clinical studies have been completed in children to assess the full reversibility of fertility suppression, animal studies (prepubertal and adult rats and monkeys) with leuprolide acetate and other GnRH analogs have shown functional recovery. However, following a study with leuprolide acetate, immature male rats demonstrated tubular degeneration in the testes even after a recovery period. In spite of the failure to recover histologically, the treated males proved to be as fertile as the controls. Also, no histologic changes were observed in the female rats following the same protocol. In both sexes, the offspring of the treated animals appeared normal. The effect of the treatment of the parents on the reproductive performance of the F1 generation was not tested. The clinical significance of these findings is unknown.
Pregnancy, Teratogenic Effects: Pregnancy Category X (see CONTRAINDICATIONS section). When administered on day 6 of pregnancy at test dosages of 0.00024, 0.0024, and 0.024 mg/kg (1/1200 to 1/12 the human pediatric dose) to rabbits, LUPRON produced a dose-related increase in major fetal abnormalities. Similar studies in rats failed to demonstrate an increase in fetal malformations. There was increased fetal mortality and decreased fetal weights with the two higher doses of LUPRON in rabbits and with the highest dose in rats.
Nursing Mothers: It is not known whether leuprolide acetate is excreted in human milk. LUPRON should not be used by nursing mothers.
Geriatric Use: See labeling for LUPRON INJECTION for the pharmacokinetics, efficacy and safety of LUPRON in this population.
ADVERSE REACTIONS
Clinical Trials:
Potential exacerbation of signs and symptoms during the first few weeks of treatment (see PRECAUTIONS section) is a concern in patients with rapidly advancing central precocious puberty.
In two studies of children with central precocious puberty, in 2% or more of the patients receiving the drug, the following adverse reactions were reported to have a possible or probable relationship to drug as ascribed by the treating physician. Reactions considered not drug related are excluded.
Number of Patients N = 395 (Percent) Body as a WholeGeneral Pain7 (2) Integumentary SystemAcne/Seborrhea7 (2) Injection Site ReactionsIncluding Abscess21 (5) Rash IncludingErythema Multiforme8 (2) Urogenital SystemVaginitis/Bleeding/Discharge7 (2)
In those same studies, the following adverse reactions were reported in less than 2% of the patients.
Body as a Whole - Body Odor, Fever, Headache, Infection; Cardiovascular System - Syncope, Vasodilation; Digestive System - Dysphagia, Gingivitis, Nausea/Vomiting; Endocrine System - Accelerated Sexual Maturity; Metabolic and Nutritional Disorders - Peripheral Edema, Weight Gain; Nervous System - Nervousness, Personality Disorder, Somnolence, Emotional Lability; Respiratory System - Epistaxis; Integumentary System - Alopecia, Skin Striae; Urogenital System - Cervix Disorder, Gynecomastia/Breast Disorders, Urinary Incontinence.
Postmarketing
During postmarketing surveillance, which includes other dosage forms and other patient populations, the following adverse events were reported.
Symptoms consistent with an anaphylactoid or asthmatic process have been rarely (incidence rate of about 0.002%) reported. Rash, urticaria, and photosensitivity reactions have also been reported. Localized reactions including induration and abscess have been reported at the site of injection. Symptoms consistent with fibromyalgia (e.g., joint and muscle pain, headaches, sleep disorders, gastrointestinal distress, and shortness of breath) have been reported individually and collectively.
Cardiovascular System - Hypotension, Pulmonary embolism; Gastrointestinal System - Hepatic dysfunction; Hemic and Lymphatic System - Decreased WBC; Integumentary System - Hair growth; Central/Peripheral Nervous System - Peripheral neuropathy, Spinal fracture/paralysis, Hearing disorder; Miscellaneous - Hard nodule in throat, Weight gain, Increased uric acid; Musculoskeletal System - Tenosynovitis-like symptoms; Respiratory System - Respiratory disorders; Urogenital System - Prostate pain.
Changes in Bone Density: Decreased bone density has been reported in the medical literature in men who have had orchiectomy or who have been treated with an LH-RH agonist analog. In a clinical trial, 25 men with prostate cancer, 12 of whom had been treated previously with leuprolide acetate for at least six months, underwent bone density studies as a result of pain. The leuprolide-treated group had lower bone density scores than the nontreated control group. The effects on bone density in children are unknown.
See other LUPRON INJECTION and LUPRON DEPOT package inserts for adverse events reported in other patient populations.
OVERDOSAGE
In rats, subcutaneous administration of 125 to 250 times the recommended human pediatric dose, expressed on a per body weight basis, resulted in dyspnea, decreased activity, and local irritation at the injection site. There is no evidence at present that there is a clinical counterpart of this phenomenon. In early clinical trials using leuprolide acetate in adult patients, doses as high as 20 mg/day for up to two years caused no adverse effects differing from those observed with the 1 mg/day dose.
DOSAGE AND ADMINISTRATION
LUPRON INJECTION can be administered by a patient/parent or health care professional.
The dose of LUPRON INJECTION must be individualized for each child. The dose is based on a mg/kg ratio of drug to body weight. Younger children require higher doses on a mg/kg ratio.
After 1-2 months of initiating therapy or changing doses, the child must be monitored with a GnRH stimulation test, sex steroids, and Tanner staging to confirm downregulation. Measurements of bone age for advancement should be monitored every 6-12 months. The dose should be titrated upward until no progression of the condition is noted either clinically and/or by laboratory parameters.
The first dose found to result in adequate downregulation can probably be maintained for the duration of therapy in most children. However, there are insufficient data to guide dosage adjustment as patients move into higher weight categories after beginning therapy at very young ages and low dosages. It is recommended that adequate downregulation be verified in such patients whose weight has increased significantly while on therapy.
As with other drugs administered by injection, the injection site should be varied periodically.
Discontinuation of LUPRON INJECTION should be considered before age 11 for females and age 12 for males.
The recommended starting dose is 50 mcg/kg/day administered as a single subcutaneous injection. If total downregulation is not achieved, the dose should be titrated upward by 10 mcg/kg/day. This dose will be considered the maintenance dose.
Follow the pictorial directions on the reverse side of this package insert for administration.
NOTE: As with other parenteral products, inspect the solution for discoloration and particulate matter before each use.
HOW SUPPLIED
LUPRON INJECTION (leuprolide acetate) is a sterile solution supplied in a 2.8 mL multiple-dose vial. The vial is packaged as follows:
- 14 Day Patient Administration Kit with 14 disposable syringes and 28 alcohol swabs, NDC 0300-3612-28.
- Six-vial carton, NDC 0300-3612-24.
- Store below 77°F (25°C). Do not freeze. Protect from light; store vial in carton until use.
- Use the syringes supplied with LUPRON INJECTION. Insulin syringes may be substituted for use with LUPRON INJECTION.
U.S. Patent Nos. 4,005,063; 4,005,194.
REFERENCE
- MacLeod TL, et al. Anaphylactic reaction to synthetic luteinizing hormone-releasing hormone. Fertil Steril 1987 Sept;48(3):500-502.
Manufactured for
TAP Pharmaceuticals Inc.
Lake Forest, IL 60045, U.S.A.
By Abbott Laboratories
North Chicago, IL 60064, U.S.A.
® - Registered
(No. 3612)
03-5391-R4; Rev. November 2004
Subscribe to the "News" RSS Feed 
TOP ۞