-
Procrit for Injection (Ortho Biotech)
DESCRIPTION
Erythropoietin is a glycoprotein which stimulates red blood cell production. It is produced in the kidney and stimulates the division and differentiation of committed erythroid progenitors in the bone marrow. PROCRIT® (Epoetin alfa), a 165 amino acid glycoprotein manufactured by recombinant DNA technology, has the same biological effects as endogenous erythropoietin. 1 It has a molecular weight of 30,400 daltons and is produced by mammalian cells into which the human erythropoietin gene has been introduced. The product contains the identical amino acid sequence of isolated natural erythropoietin.
PROCRIT® is formulated as a sterile, colorless liquid in an isotonic sodium chloride/sodium citrate buffered solution or a sodium chloride/sodium phosphate buffered solution for intravenous (IV) or subcutaneous (SC) administration.
Single-dose, Preservative-free Vial: Each 1 mL of solution contains 2000, 3000, 4000 or 10,000 Units of Epoetin alfa, 2.5 mg Albumin (Human), 5.8 mg sodium citrate, 5.8 mg sodium chloride, and 0.06 mg citric acid in Water for Injection, USP (pH 6.9 ± 0.3). This formulation contains no preservative.
Single-dose, Preservative-free Vial: 1 mL (40,000 Units/mL). Each 1 mL of solution contains 40,000 Units of Epoetin alfa, 2.5 mg Albumin (Human), 1.2 mg sodium phosphate monobasic monohydrate, 1.8 mg sodium phosphate dibasic anhydrate, 0.7 mg sodium citrate, 5.8 mg sodium chloride, and 6.8 mcg citric acid in Water for Injection, USP (pH 6.9 ± 0.3). This formulation contains no preservative.
Multidose, Preserved Vial: 2 mL (20,000 Units, 10,000 Units/mL). Each 1 mL of solution contains 10,000 Units of Epoetin alfa, 2.5 mg Albumin (Human), 1.3 mg sodium citrate, 8.2 mg sodium chloride, 0.11 mg citric acid, and 1% benzyl alcohol as preservative in Water for Injection, USP (pH 6.1 ± 0.3).
Multidose, Preserved Vial: 1 mL (20,000 Units/mL). Each 1 mL of solution contains 20,000 Units of Epoetin alfa, 2.5 mg Albumin (Human), 1.3 mg sodium citrate, 8.2 mg sodium chloride, 0.11 mg citric acid, and 1% benzyl alcohol as preservative in Water for Injection, USP (pH 6.1 ± 0.3).
CLINICAL PHARMACOLOGY
Chronic Renal Failure Patients
Endogenous production of erythropoietin is normally regulated by the level of tissue oxygenation. Hypoxia and anemia generally increase the production of erythropoietin, which in turn stimulates erythropoiesis. 2 In normal subjects, plasma erythropoietin levels range from 0.01 to 0.03 Units/mL and increase up to 100- to 1000-fold during hypoxia or anemia. 2 In contrast, in patients with chronic renal failure (CRF), production of erythropoietin is impaired, and this erythropoietin deficiency is the primary cause of their anemia. 3,4
Chronic renal failure is the clinical situation in which there is a progressive and usually irreversible decline in kidney function. Such patients may manifest the sequelae of renal dysfunction, including anemia, but do not necessarily require regular dialysis. Patients with end-stage renal disease (ESRD) are those patients with CRF who require regular dialysis or kidney transplantation for survival.
PROCRIT® has been shown to stimulate erythropoiesis in anemic patients with CRF, including both patients on dialysis and those who do not require regular dialysis. 4-13 The first evidence of a response to the three times weekly (TIW) administration of PROCRIT® is an increase in the reticulocyte count within 10 days, followed by increases in the red cell count, hemoglobin, and hematocrit, usually within 2 to 6 weeks. 4,5 Because of the length of time required for erythropoiesis - several days for erythroid progenitors to mature and be released into the circulation - a clinically significant increase in hematocrit is usually not observed in less than 2 weeks and may require up to 6 weeks in some patients. Once the hematocrit reaches the suggested target range (30% to 36%), that level can be sustained by PROCRIT® therapy in the absence of iron deficiency and concurrent illnesses.
The rate of hematocrit increase varies between patients and is dependent upon the dose of PROCRIT®, within a therapeutic range of approximately 50 to 300 Units/kg TIW. 4 A greater biologic response is not observed at doses exceeding 300 Units/kg TIW. 6 Other factors affecting the rate and extent of response include availability of iron stores, the baseline hematocrit, and the presence of concurrent medical problems.
Zidovudine-treated HIV-infected Patients
Responsiveness to PROCRIT® in HIV-infected patients is dependent upon the endogenous serum erythropoietin level prior to treatment. Patients with endogenous serum erythropoietin levels </= 500 mUnits/mL, and who are receiving a dose of zidovudine </= 4200 mg/week, may respond to PROCRIT® therapy. Patients with endogenous serum erythropoietin levels > 500 mUnits/mL do not appear to respond to PROCRIT® therapy. In a series of four clinical trials involving 255 patients, 60% to 80% of HIV-infected patients treated with zidovudine had endogenous serum erythropoietin levels </= 500 mUnits/mL.
Response to PROCRIT® in zidovudine-treated HIV-infected patients is manifested by reduced transfusion requirements and increased hematocrit.
Cancer Patients on Chemotherapy
A series of clinical trials enrolled 131 anemic cancer patients who received PROCRIT® TIW and who were receiving cyclic cisplatin- or non cisplatin-containing chemotherapy. Endogenous baseline serum erythropoietin levels varied among patients in these trials with approximately 75% (n = 83/110) having endogenous serum erythropoietin levels </= 132 mUnits/mL, and approximately 4% (n = 4/110) of patients having endogenous serum erythropoietin levels > 500 mUnits/mL. In general, patients with lower baseline serum erythropoietin levels responded more vigorously to PROCRIT® than patients with higher baseline erythropoietin levels. Although no specific serum erythropoietin level can be stipulated above which patients would be unlikely to respond to PROCRIT® therapy, treatment of patients with grossly elevated serum erythropoietin levels (eg, > 200 mUnits/mL) is not recommended.
Pharmacokinetics
In adult and pediatric patients with CRF, the elimination half-life of plasma erythropoietin after intravenously administered PROCRIT® ranges from 4 to 13 hours. 14-16 The half-life is approximately 20% longer in CRF patients than that in healthy subjects. After SC administration, peak plasma levels are achieved within 5 to 24 hours. The half-life is similar between adult patients with serum creatinine level greater than 3 and not on dialysis and those maintained on dialysis.
The pharmacokinetic data indicate no apparent difference in PROCRIT® half-life among adult patients above or below 65 years of age.The pharmacokinetic profile of PROCRIT® in children and adolescents appears to be similar to that of adults. Limited data are available in neonates. 17
A study of 7 preterm very low birth weight neonates and 10 healthy adults given IV erythropoietin suggested that distribution volume was approximately 1.5 to 2 times higher in the preterm neonates than in the healthy adults, and clearance was approximately 3 times higher in the preterm neonates than in the healthy adults. 44
The pharmacokinetics of PROCRIT® have not been studied in HIV-infected patients.
A pharmacokinetic study comparing 150 Units/kg SC TIW to 40,000 Units SC weekly dosing regimen was conducted for 4 weeks in healthy subjects (n = 12) and for 6 weeks in anemic cancer patients (n = 32) receiving cyclic chemotherapy. There was no accumulation of serum erythropoietin after the 2 dosing regimens during the study period. The 40,000 Units weekly regimen had a higher C max (3- to 7-fold), longer T max (2- to 3-fold), higher AUC 0-168h (2- to 3-fold) of erythropoietin and lower clearance (50%) than the 150 Units/kg TIW regimen. In anemic cancer patients, the average t 1/2 was similar (40 hours with range of 16 to 67 hours) after both dosing regimens. After the 150 Units/kg TIW dosing, the values of T max and clearance are similar (13.3 ± 12.4 vs. 14.2 ± 6.7 hours, and 20.2 ± 15.9 vs. 23.6 ± 9.5 mL/h/kg) between Week 1 when patients were receiving chemotherapy (n = 14) and Week 3 when patients were not receiving chemotherapy (n = 4). Differences were observed after the 40,000 Units weekly dosing with longer T max (38 ± 18 hours) and lower clearance (9.2 ± 4.7 mL/h/kg) during Week 1 when patients were receiving chemotherapy (n = 18) compared with those (22 ± 4.5 hours, 13.9 ± 7.6 mL/h/kg) during Week 3 when patients were not receiving chemotherapy (n = 7).
The bioequivalence between the 10,000 Units/mL citrate-buffered Epoetin alfa formulation and the 40,000 Units/mL phosphate-buffered Epoetin alfa formulation has been demonstrated after SC administration of single 750 Units/kg doses to healthy subjects.
INDICATIONS AND USAGE
Treatment of Anemia of Chronic Renal Failure Patients
PROCRIT® is indicated for the treatment of anemia associated with CRF, including patients on dialysis (ESRD) and patients not on dialysis. PROCRIT® is indicated to elevate or maintain the red blood cell level (as manifested by the hematocrit or hemoglobin determinations) and to decrease the need for transfusions in these patients.
Non-dialysis patients with symptomatic anemia considered for therapy should have a hemoglobin less than 10 g/dL.
PROCRIT® is not intended for patients who require immediate correction of severe anemia. PROCRIT® may obviate the need for maintenance transfusions but is not a substitute for emergency transfusion.
Prior to initiation of therapy, the patient's iron stores should be evaluated. Transferrin saturation should be at least 20% and ferritin at least 100 ng/mL. Blood pressure should be adequately controlled prior to initiation of PROCRIT® therapy, and must be closely monitored and controlled during therapy.
PROCRIT® should be administered under the guidance of a qualified physician (see DOSAGE AND ADMINISTRATION ).
Treatment of Anemia in Zidovudine-treated HIV-infected Patients
PROCRIT® is indicated for the treatment of anemia related to therapy with zidovudine in HIV-infected patients. PROCRIT® is indicated to elevate or maintain the red blood cell level (as manifested by the hematocrit or hemoglobin determinations) and to decrease the need for transfusions in these patients. PROCRIT® is not indicated for the treatment of anemia in HIV-infected patients due to other factors such as iron or folate deficiencies, hemolysis, or gastrointestinal bleeding, which should be managed appropriately.
PROCRIT®, at a dose of 100 Units/kg TIW, is effective in decreasing the transfusion requirement and increasing the red blood cell level of anemic, HIV-infected patients treated with zidovudine, when the endogenous serum erythropoietin level is </= 500 mUnits/mL and when patients are receiving a dose of zidovudine </= 4200 mg/week.
Treatment of Anemia in Cancer Patients on Chemotherapy
PROCRIT® is indicated for the treatment of anemia in patients with non-myeloid malignancies where anemia is due to the effect of concomitantly administered chemotherapy. PROCRIT® is indicated to decrease the need for transfusions in patients who will be receiving concomitant chemotherapy for a minimum of 2 months. PROCRIT® is not indicated for the treatment of anemia in cancer patients due to other factors such as iron or folate deficiencies, hemolysis, or gastrointestinal bleeding, which should be managed appropriately.
Reduction of Allogeneic Blood Transfusion in Surgery Patients
PROCRIT® is indicated for the treatment of anemic patients (hemoglobin > 10 to </= 13 g/dL) scheduled to undergo elective, noncardiac, nonvascular surgery to reduce the need for allogeneic blood transfusions. 18-20 PROCRIT® is indicated for patients at high risk for perioperative transfusions with significant, anticipated blood loss. PROCRIT® is not indicated for anemic patients who are willing to donate autologous blood. The safety of the perioperative use of PROCRIT® has been studied only in patients who are receiving anticoagulant prophylaxis.
CLINICAL EXPERIENCE: RESPONSE TO PROCRIT®
Chronic Renal Failure Patients
Response to PROCRIT® was consistent across all studies. In the presence of adequate iron stores (see IRON EVALUATION ), the time to reach the target hematocrit is a function of the baseline hematocrit and the rate of hematocrit rise.
The rate of increase in hematocrit is dependent upon the dose of PROCRIT® administered and individual patient variation. In clinical trials at starting doses of 50 to 150 Units/kg TIW, adult patients responded with an average rate of hematocrit rise of:
Starting Dose
(TIW IV)Hematocrit Increase Points/Day Points/2 Weeks 50 Units/kg 0.11 1.5 100 Units/kg 0.18 2.5 150 Units/kg 0.25 3.5 Over this dose range, approximately 95% of all patients responded with a clinically significant increase in hematocrit, and by the end of approximately 2 months of therapy virtually all patients were transfusion-independent. Changes in the quality of life of adult patients treated with PROCRIT® were assessed as part of a phase 3 clinical trial. 5,8 Once the target hematocrit (32% to 38%) was achieved, statistically significant improvements were demonstrated for most quality of life parameters measured, including energy and activity level, functional ability, sleep and eating behavior, health status, satisfaction with health, sex life, well-being, psychological effect, life satisfaction, and happiness. Patients also reported improvement in their disease symptoms. They showed a statistically significant increase in exercise capacity (VO 2 max), energy, and strength with a significant reduction in aching, dizziness, anxiety, shortness of breath, muscle weakness, and leg cramps. 8,21
Adult Patients on Dialysis: Thirteen clinical studies were conducted, involving IV administration to a total of 1010 anemic patients on dialysis for 986 patient-years of PROCRIT® therapy. In the three largest of these clinical trials, the median maintenance dose necessary to maintain the hematocrit between 30% to 36% was approximately 75 Units/kg TIW. In the US multicenter phase 3 study, approximately 65% of the patients required doses of 100 Units/kg TIW, or less, to maintain their hematocrit at approximately 35%. Almost 10% of patients required a dose of 25 Units/kg, or less, and approximately 10% required a dose of more than 200 Units/kg TIW to maintain their hematocrit at this level.
A multicenter unit dose study was also conducted in 119 patients receiving peritoneal dialysis who self-administered PROCRIT® subcutaneously for approximately 109 patient-years of experience. Patients responded to PROCRIT® administered SC in a manner similar to patients receiving IV administration. 22
Pediatric Patients on Dialysis: One hundred twenty-eight children from 2 months to 19 years of age with CRF requiring dialysis were enrolled in 4 clinical studies of PROCRIT®. The largest study was a placebo-controlled, randomized trial in 113 children with anemia (hematocrit </= 27%) undergoing peritoneal dialysis or hemodialysis. The initial dose of PROCRIT® was 50 Units/kg IV or SC TIW. The dose of study drug was titrated to achieve either a hematocrit of 30% to 36% or an absolute increase in hematocrit of 6 percentage points over baseline.
At the end of the initial 12 weeks, a statistically significant rise in mean hematocrit (9.4% vs 0.9%) was observed only in the PROCRIT® arm. The proportion of children achieving a hematocrit of 30%, or an increase in hematocrit of 6 percentage points over baseline, at any time during the first 12 weeks was higher in the PROCRIT® arm (96% vs 58%). Within 12 weeks of initiating PROCRIT® therapy, 92.3% of the pediatric patients were transfusion-independent as compared to 65.4% who received placebo. Among patients who received 36 weeks of PROCRIT®, hemodialysis patients required a higher median maintenance dose (167 Units/kg/week [n = 28] vs 76 Units/kg/week [n = 36]) and took longer to achieve a hematocrit of 30% to 36% (median time to response 69 days vs 32 days) than patients undergoing peritoneal dialysis.
Patients With CRF Not Requiring Dialysis
Four clinical trials were conducted in patients with CRF not on dialysis involving 181 patients treated with PROCRIT® for approximately 67 patient-years of experience. These patients responded to PROCRIT® therapy in a manner similar to that observed in patients on dialysis. Patients with CRF not on dialysis demonstrated a dose-dependent and sustained increase in hematocrit when PROCRIT® was administered by either an IV or SC route, with similar rates of rise of hematocrit when PROCRIT® was administered by either route. Moreover, PROCRIT® doses of 75 to 150 Units/kg per week have been shown to maintain hematocrits of 36% to 38% for up to 6 months. Correcting the anemia of progressive renal failure will allow patients to remain active even though their renal function continues to decrease. 23-24
Zidovudine-treated HIV-infected Patients
PROCRIT® has been studied in four placebo-controlled trials enrolling 297 anemic (hematocrit < 30%) HIV-infected (AIDS) patients receiving concomitant therapy with zidovudine (all patients were treated with Epoetin alfa manufactured by Amgen Inc). In the subgroup of patients (89/125 PROCRIT® and 88/130 placebo) with prestudy endogenous serum erythropoietin levels </= 500 mUnits/mL, PROCRIT® reduced the mean cumulative number of units of blood transfused per patient by approximately 40% as compared to the placebo group. 25 Among those patients who required transfusions at baseline, 43% of patients treated with PROCRIT® versus 18% of placebo-treated patients were transfusion-independent during the second and third months of therapy. PROCRIT® therapy also resulted in significant increases in hematocrit in comparison to placebo. When examining the results according to the weekly dose of zidovudine received during month 3 of therapy, there was a statistically significant (p < 0.003) reduction in transfusion requirements in patients treated with PROCRIT® (n = 51) compared to placebo treated patients (n = 54) whose mean weekly zidovudine dose was </= 4200 mg/week. 25
Approximately 17% of the patients with endogenous serum erythropoietin levels </= 500 mUnits/mL receiving PROCRIT® in doses from 100 to 200 Units/kg TIW achieved a hematocrit of 38% without administration of transfusions or significant reduction in zidovudine dose. In the subgroup of patients whose prestudy endogenous serum erythropoietin levels were > 500 mUnits/mL, PROCRIT® therapy did not reduce transfusion requirements or increase hematocrit, compared to the corresponding responses in placebo-treated patients.
In a 6 month open-label PROCRIT® study, patients responded with decreased transfusion requirements and sustained increases in hematocrit and hemoglobin with doses of PROCRIT® up to 300 Units/kg TIW. 25-27
Responsiveness to PROCRIT® therapy may be blunted by intercurrent infectious/inflammatory episodes and by an increase in zidovudine dosage. Consequently, the dose of PROCRIT® must be titrated based on these factors to maintain the desired erythropoietic response.
Cancer Patients on Chemotherapy
Three-Times Weekly (TIW) Dosing
PROCRIT® administered TIW has been studied in a series of six placebo-controlled, double-blind trials that enrolled 131 anemic cancer patients receiving PROCRIT® or matching placebo. Across all studies, 72 patients were treated with concomitant non cisplatin-containing chemotherapy regimens and 59 patients were treated with concomitant cisplatin-containing chemotherapy regimens. Patients were randomized to PROCRIT® 150 Units/kg or placebo subcutaneously TIW for 12 weeks in each study.
The results of the pooled data from these six studies are shown in the table below. Because of the length of time required for erythropoiesis and red cell maturation, the efficacy of PROCRIT® (reduction in proportion of patients requiring transfusions) is not manifested until 2 to 6 weeks after initiation of PROCRIT®.
Proportion of Patients Transfused During Chemotherapy
(Efficacy Population a )Chemotherapy Regimen On Study b During Months 2 and 3 c Regimens
without cisplatinPROCRIT® Placebo PROCRIT® Placebo 44% (15/34) 44% (16/36) 21% (6/29) 33% (11/33) Regimens
containing cisplatin50% (14/28) 63% (19/30) 23% (5/22) d 56% (14/25) Combined47% (29/62) 53% (35/66) 22% (11/51) d 43% (25/58) a Limited to patients remaining on study at least 15 days (1 patient excluded from PROCRIT®, 2 patients excluded from placebo). b Includes all transfusions from day 1 through the end of study. c Limited to patients remaining on study beyond week 6 and includes only transfusions during weeks 5-12. d Unadjusted 2-sided p < 0.05. Intensity of chemotherapy in the above trials was not directly assessed, however the degree and timing of neutropenia was comparable across all trials. Available evidence suggests that patients with lymphoid and solid cancers respond similarly to PROCRIT® therapy, and that patients with or without tumor infiltration of the bone marrow respond similarly to PROCRIT® therapy.
Weekly (QW) Dosing
PROCRIT® was also studied in a placebo-controlled, double-blind trial utilizing weekly dosing in a total of 344 anemic cancer patients. In this trial, 61 (35 placebo arm and 26 in the PROCRIT® arm) patients were treated with concomitant cisplatin containing regimens and 283 patients received concomitant chemotherapy regimens that did not contain cisplatinum. Patients were randomized to PROCRIT® 40,000 Units weekly (n = 174) or placebo (n = 170) SC for a planned treatment period of 16 weeks. If hemoglobin had not increased by > 1 g/dL, after 4 weeks of therapy or the patient received RBC transfusion during the first 4 weeks of therapy, study drug was increased to 60,000 Units weekly. Forty-three percent of patients in the Epoetin alfa group required an increase in PROCRIT® dose to 60,000 Units weekly. 25
Results demonstrated that PROCRIT® therapy reduced the proportion of patients transfused in day 29 through week 16 of the study as compared to placebo. Twenty-five patients (14%) in the PROCRIT® group received transfusions compared to 48 patients (28%) in the placebo group (p = 0.0010) between day 29 and week 16 or the last day on study.
Comparable intensity of chemotherapy for patients enrolled in the two study arms was suggested by similarities in mean dose and frequency of administration for the 10 most commonly administered chemotherapy agents, and similarity in the incidence of changes in chemotherapy during the trial in the two arms.
Surgery Patients
PROCRIT® has been studied in a placebo-controlled, double-blind trial enrolling 316 patients scheduled for major, elective orthopedic hip or knee surgery who were expected to require >/= 2 units of blood and who were not able or willing to participate in an autologous blood donation program. Based on previous studies which demonstrated that pretreatment hemoglobin is a predictor of risk of receiving transfusion, 20,28 patients were stratified into one of three groups based on their pretreatment hemoglobin [</= 10 (n = 2), > 10 to </= 13 (n = 96), and > 13 to </= 15 g/dL (n = 218)] and then randomly assigned to receive 300 Units/kg PROCRIT®, 100 Units/kg PROCRIT® or placebo by SC injection for 10 days before surgery, on the day of surgery, and for 4 days after surgery. 18 All patients received oral iron and a low-dose post-operative warfarin regimen. 18
Treatment with PROCRIT® 300 Units/kg significantly (p = 0.024) reduced the risk of allogeneic transfusion in patients with a pretreatment hemoglobin of > 10 to </= 13; 5/31 (16%) of PROCRIT® 300 Units/kg, 6/26 (23%) of PROCRIT® 100 Units/kg, and 13/29 (45%) of placebo-treated patients were transfused. 18 There was no significant difference in the number of patients transfused between PROCRIT® (9% 300 Units/kg, 6% 100 Units/kg) and placebo (13%) in the > 13 to </= 15 g/dL hemoglobin stratum. There were too few patients in the </= 10 g/dL group to determine if PROCRIT® is useful in this hemoglobin strata. In the > 10 to </= 13 g/dL pretreatment stratum, the mean number of units transfused per PROCRIT®-treated patient (0.45 units blood for 300 Units/kg, 0.42 units blood for 100 Units/kg) was less than the mean transfused per placebo-treated patient (1.14 units) (overall p = 0.028). In addition, mean hemoglobin, hematocrit, and reticulocyte counts increased significantly during the presurgery period in patients treated with PROCRIT®. 18
PROCRIT® was also studied in an open-label, parallel-group trial enrolling 145 subjects with a pretreatment hemoglobin level of >/= 10 to </= 13 g/dL who were scheduled for major orthopedic hip or knee surgery and who were not participating in an autologous program. 19 Subjects were randomly assigned to receive one of two SC dosing regimens of PROCRIT® (600 Units/kg once weekly for 3 weeks prior to surgery and on the day of surgery, or 300 Units/kg once daily for 10 days prior to surgery, on the day of surgery and for 4 days after surgery). All subjects received oral iron and appropriate pharmacologic anticoagulation therapy.
From pretreatment to presurgery, the mean increase in hemoglobin in the 600 Units/kg weekly group (1.44 g/dL) was greater than observed in the 300 Units/kg daily group. 19 The mean increase in absolute reticulocyte count was smaller in the weekly group (0.11 × 10 6 /mm 3 ) compared to the daily group (0.17 × 10 6 /mm 3 ). Mean hemoglobin levels were similar for the two treatment groups throughout the postsurgical period.
The erythropoietic response observed in both treatment groups resulted in similar transfusion rates [11/69 (16%) in the 600 Units/kg weekly group and 14/71 (20%) in the 300 Units/kg daily group]. 19 The mean number of units transfused per subject was approximately 0.3 units in both treatment groups.
CONTRAINDICATIONS
PROCRIT® is contraindicated in patients with:
- Uncontrolled hypertension.
- Known hypersensitivity to mammalian cell-derived products.
- Known hypersensitivity to Albumin (Human).
WARNINGS
Pediatric Use
The multidose preserved formulation contains benzyl alcohol. Benzyl alcohol has been reported to be associated with an increased incidence of neurological and other complications in premature infants which are sometimes fatal.
Thrombotic Events and Increased Mortality
A randomized, prospective trial of 1265 hemodialysis patients with clinically evident cardiac disease (ischemic heart disease or congestive heart failure) was conducted in which patients were assigned to PROCRIT® treatment targeted to a maintenance hematocrit of either 42 ± 3% or 30 ± 3%. 42 Increased mortality was observed in 634 patients randomized to a target hematocrit of 42% [221 deaths (35% mortality)] compared to 631 patients targeted to remain at a hematocrit of 30% [185 deaths (29% mortality)]. The reason for the increased mortality observed in these studies is unknown, however, the incidence of non-fatal myocardial infarctions (3.1% vs 2.3%), vascular access thromboses (39% vs 29%), and all other thrombotic events (22% vs 18%) were also higher in the group randomized to achieve a hematocrit of 42%.
Increased mortality was also observed in a randomized placebo-controlled study of PROCRIT® in adult patients who did not have CRF who were undergoing coronary artery bypass surgery (7 deaths in 126 patients randomized to PROCRIT® versus no deaths among 56 patients receiving placebo). Four of these deaths occurred during the period of study drug administration and all four deaths were associated with thrombotic events. While the extent of the population affected is unknown, in patients at risk for thrombosis, the anticipated benefits of PROCRIT® treatment should be weighed against the potential for increased risks associated with therapy.
In a randomized, prospective trial conducted with another Epoetin alfa product, in 939 women with metastatic carcinoma of the breast who were receiving chemotherapy, patients were assigned to receive either Epoetin alfa or placebo for up to a year, in a weekly schedule, with the primary goal of showing improved survival and improved quality of life in the Epoetin alfa treatment arm. 25 This study utilized a treatment strategy designed to maintain hemoglobin levels of 12 to 14 g/dL (hematocrit 36 to 42%). Increased mortality in the first 4 months after randomization was observed among 469 patients who received the erythropoietin product [41 deaths (8.7% mortality)] compared to 470 patients who received placebo [16 deaths (3.4% mortality)]. In the first four months of the study, the incidence of fatal thrombotic vascular events (1.1% vs 0.2%) and death attributed to disease progression (6.0% vs 2.8%) were both higher in the group randomized to receive Epoetin alfa as compared to placebo. Based on Kaplan-Meier estimates, the proportion of subjects surviving at 12 months after randomization was lower in the Epoetin alfa group than in the placebo group (70% vs 76%), p = 0.012, log rank. However, due to insufficient monitoring and data collection, reliable comparisons cannot be made concerning the effect of Epoetin alfa on overall time to disease progression, progression-free survival, and overall survival.
Pure Red Cell Aplasia
Pure red cell aplasia (PRCA), in association with neutralizing antibodies to native erythropoietin, has been observed in patients treated with recombinant erythropoietins. PRCA has been reported in a limited number of patients exposed to PROCRIT®. This has been reported predominantly in patients with CRF. Any patient with loss of response to PROCRIT® should be evaluated for the etiology of loss of effect (see PRECAUTIONS : LACK OR LOSS OF RESPONSE ). PROCRIT® should be discontinued in any patient with evidence of PRCA and the patient evaluated for the presence of binding and neutralizing antibodies to PROCRIT®, native erythropoietin, and any other recombinant erythropoietin administered to the patient. Amgen/Ortho Biotech Products, L.P. should be contacted to assist in this evaluation. In patients with PRCA secondary to neutralizing antibodies to erythropoietin, PROCRIT® should not be administered and such patients should not be switched to another product as anti-erythropoietin antibodies cross-react with other erythropoietins (see ADVERSE REACTIONS ).
Albumin (Human)
PROCRIT® contains albumin, a derivative of human blood. Based on effective donor screening and product manufacturing processes, it carries an extremely remote risk for transmission of viral diseases. A theoretical risk for transmission of Creutzfeldt-Jakob disease (CJD) also is considered extremely remote. No cases of transmission of viral diseases or CJD have ever been identified for albumin.
Chronic Renal Failure Patients
Hypertension: Patients with uncontrolled hypertension should not be treated with PROCRIT®; blood pressure should be controlled adequately before initiation of therapy. Up to 80% of patients with CRF have a history of hypertension. 29 Although there does not appear to be any direct pressor effects of PROCRIT®, blood pressure may rise during PROCRIT® therapy. During the early phase of treatment when the hematocrit is increasing, approximately 25% of patients on dialysis may require initiation of, or increases in, antihypertensive therapy. Hypertensive encephalopathy and seizures have been observed in patients with CRF treated with PROCRIT®.
Special care should be taken to closely monitor and aggressively control blood pressure in patients treated with PROCRIT® . Patients should be advised as to the importance of compliance with antihypertensive therapy and dietary restrictions. If blood pressure is difficult to control by initiation of appropriate measures, the hemoglobin may be reduced by decreasing or withholding the dose of PROCRIT®. A clinically significant decrease in hemoglobin may not be observed for several weeks.
It is recommended that the dose of PROCRIT® be decreased if the hemoglobin increase exceeds 1 g/dL in any 2-week period, because of the possible association of excessive rate of rise of hemoglobin with an exacerbation of hypertension. In CRF patients on hemodialysis with clinically evident ischemic heart disease or congestive heart failure, the hemoglobin should be managed carefully, not to exceed 12 g/dL (see THROMBOTIC EVENTS ).
Seizures: Seizures have occurred in patients with CRF participating in PROCRIT® clinical trials.
In adult patients on dialysis, there was a higher incidence of seizures during the first 90 days of therapy (occurring in approximately 2.5% of patients) as compared with later timepoints.
Given the potential for an increased risk of seizures during the first 90 days of therapy, blood pressure and the presence of premonitory neurologic symptoms should be monitored closely. Patients should be cautioned to avoid potentially hazardous activities such as driving or operating heavy machinery during this period.
While the relationship between seizures and the rate of rise of hemoglobin is uncertain, it is recommended that the dose of PROCRIT® be decreased if the hemoglobin increase exceeds 1 g/dL in any 2-week period.
Thrombotic Events: During hemodialysis, patients treated with PROCRIT® may require increased anticoagulation with heparin to prevent clotting of the artificial kidney (see ADVERSE REACTIONS for more information about thrombotic events).
Other thrombotic events (eg, myocardial infarction, cerebrovascular accident, transient ischemic attack) have occurred in clinical trials at an annualized rate of less than 0.04 events per patient year of PROCRIT® therapy. These trials were conducted in adult patients with CRF (whether on dialysis or not) in whom the target hematocrit was 32% to 40%. However, the risk of thrombotic events, including vascular access thrombosis, was significantly increased in adult patients with ischemic heart disease or congestive heart failure receiving PROCRIT® therapy with the goal of reaching a normal hematocrit (42%) as compared to a target hematocrit of 30%. Patients with pre-existing cardiovascular disease should be monitored closely.
Zidovudine-treated HIV-infected Patients
In contrast to CRF patients, PROCRIT® therapy has not been linked to exacerbation of hypertension, seizures, and thrombotic events in HIV-infected patients.
PRECAUTIONS
The parenteral administration of any biologic product should be attended by appropriate precautions in case allergic or other untoward reactions occur (see CONTRAINDICATIONS ). In clinical trials, while transient rashes were occasionally observed concurrently with PROCRIT® therapy, no serious allergic or anaphylactic reactions were reported (see ADVERSE REACTIONS for more information regarding allergic reactions).
The safety and efficacy of PROCRIT® therapy have not been established in patients with a known history of a seizure disorder or underlying hematologic disease (eg, sickle cell anemia, myelodysplastic syndromes, or hypercoagulable disorders).
In some female patients, menses have resumed following PROCRIT® therapy; the possibility of pregnancy should be discussed and the need for contraception evaluated.
Hematology
Exacerbation of porphyria has been observed rarely in patients with CRF treated with PROCRIT®. However, PROCRIT® has not caused increased urinary excretion of porphyrin metabolites in normal volunteers, even in the presence of a rapid erythropoietic response. Nevertheless, PROCRIT® should be used with caution in patients with known porphyria.
In preclinical studies in dogs and rats, but not in monkeys, PROCRIT® therapy was associated with subclinical bone marrow fibrosis. Bone marrow fibrosis is a known complication of CRF in humans and may be related to secondary hyperparathyroidism or unknown factors. The incidence of bone marrow fibrosis was not increased in a study of adult patients on dialysis who were treated with PROCRIT® for 12 to 19 months, compared to the incidence of bone marrow fibrosis in a matched group of patients who had not been treated with PROCRIT®.
Hemoglobin in CRF patients should be measured twice a week; zidovudine-treated HIV-infected and cancer patients should have hemoglobin measured once a week until hemoglobin has been stabilized, and measured periodically thereafter.
Lack or Loss of Response
If the patient fails to respond or to maintain a response to doses within the recommended dosing range, the following etiologies should be considered and evaluated:
- Iron deficiency: Virtually all patients will eventually require supplemental iron therapy (see IRON EVALUATION ).
- Underlying infectious, inflammatory, or malignant processes.
- Occult blood loss.
- Underlying hematologic diseases (ie, thalassemia, refractory anemia, or other myelodysplastic disorders).
- Vitamin deficiencies: Folic acid or vitamin B12.
- Hemolysis.
- Aluminum intoxication.
- Osteitis fibrosa cystica.
- Pure Red Cell Aplasia (PRCA): In the absence of another etiology, the patient should be evaluated for evidence of PRCA and sera should be tested for the presence of antibodies to recombinant erythropoietins.
Iron Evaluation
During PROCRIT® therapy, absolute or functional iron deficiency may develop. Functional iron deficiency, with normal ferritin levels but low transferrin saturation, is presumably due to the inability to mobilize iron stores rapidly enough to support increased erythropoiesis. Transferrin saturation should be at least 20% and ferritin should be at least 100 ng/mL.
Prior to and during PROCRIT® therapy, the patient's iron status, including transferrin saturation (serum iron divided by iron binding capacity) and serum ferritin, should be evaluated. Virtually all patients will eventually require supplemental iron to increase or maintain transferrin saturation to levels which will adequately support erythropoiesis stimulated by PROCRIT®. All surgery patients being treated with PROCRIT® should receive adequate iron supplementation throughout the course of therapy in order to support erythropoiesis and avoid depletion of iron stores.
Drug Interactions
No evidence of interaction of PROCRIT® with other drugs was observed in the course of clinical trials.
Carcinogenesis, Mutagenesis, and Impairment of Fertility
Carcinogenic potential of PROCRIT® has not been evaluated. PROCRIT® does not induce bacterial gene mutation (Ames Test), chromosomal aberrations in mammalian cells, micronuclei in mice, or gene mutation at the HGPRT locus. In female rats treated IV with PROCRIT®, there was a trend for slightly increased fetal wastage at doses of 100 and 500 Units/kg.
Pregnancy Category C
PROCRIT® has been shown to have adverse effects in rats when given in doses 5 times the human dose. There are no adequate and well-controlled studies in pregnant women. PROCRIT® should be used during pregnancy only if potential benefit justifies the potential risk to the fetus.
In studies in female rats, there were decreases in body weight gain, delays in appearance of abdominal hair, delayed eyelid opening, delayed ossification, and decreases in the number of caudal vertebrae in the F1 fetuses of the 500 Units/kg group. In female rats treated IV, there was a trend for slightly increased fetal wastage at doses of 100 and 500 Units/kg. PROCRIT® has not shown any adverse effect at doses as high as 500 Units/kg in pregnant rabbits (from day 6 to 18 of gestation).
Nursing Mothers
Postnatal observations of the live offspring (F1 generation) of female rats treated with PROCRIT® during gestation and lactation revealed no effect of PROCRIT® at doses of up to 500 Units/kg. There were, however, decreases in body weight gain, delays in appearance of abdominal hair, eyelid opening, and decreases in the number of caudal vertebrae in the F1 fetuses of the 500 Units/kg group. There were no PROCRIT®-related effects on the F2 generation fetuses.
It is not known whether PROCRIT® is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when PROCRIT® is administered to a nursing woman.
Pediatric Use
See WARNINGS : PEDIATRIC USE .
Pediatric Patients on Dialysis: PROCRIT® is indicated in infants (1 month to 2 years), children (2 years to 12 years), and adolescents (12 years to 16 years) for the treatment of anemia associated with CRF requiring dialysis. Safety and effectiveness in pediatric patients less than 1 month old have not been established (see CLINICAL EXPERIENCE: CHRONIC RENAL FAILURE , PEDIATRIC PATIENTS ON DIALYSIS ). The safety data from these studies show that there is no increased risk to pediatric CRF patients on dialysis when compared to the safety profile of PROCRIT® in adult CRF patients (see ADVERSE REACTIONS and WARNINGS ). Published literature 30-33 provides supportive evidence of the safety and effectiveness of PROCRIT® in pediatric CRF patients on dialysis.
Pediatric Patients Not Requiring Dialysis: Published literature 33,34 has reported the use of PROCRIT® in 133 pediatric patients with anemia associated with CRF not requiring dialysis, ages 3 months to 20 years, treated with 50 to 250 Units/kg SC or IV, QW to TIW. Dose-dependent increases in hemoglobin and hematocrit were observed with reductions in transfusion requirements.
Pediatric HIV-infected Patients: Published literature 35,36 has reported the use of PROCRIT® in 20 zidovudine-treated anemic HIV-infected pediatric patients ages 8 months to 17 years, treated with 50 to 400 Units/kg SC or IV, 2 to 3 times per week. Increases in hemoglobin levels and in reticulocyte counts, and decreases in or elimination of blood transfusions were observed.
Pediatric Cancer Patients on Chemotherapy: Published literature 37,38 has reported the use of PROCRIT® in approximately 64 anemic pediatric cancer patients ages 6 months to 18 years, treated with 25 to 300 Units/kg SC or IV, 3 to 7 times per week. Increases in hemoglobin and decreases in transfusion requirements were noted.
Geriatric Use
Among 1051 patients enrolled in the 5 clinical trials of PROCRIT® for reduction of allogeneic blood transfusions in patients undergoing elective surgery 745 received PROCRIT® and 306 received placebo. Of the 745 patients who received PROCRIT®, 432 (58%) were aged 65 and over, while 175 (23%) were 75 and over. No overall differences in safety or effectiveness were observed between geriatric and younger patients. The dose requirements for PROCRIT® in geriatric and younger patients within the 4 trials using the TIW schedule were similar. Insufficient numbers of patients were enrolled in the study using the weekly dosing regimen to determine whether the dosing requirements differ for this schedule.
Of the 882 patients enrolled in the 3 studies of chronic renal failure patients on dialysis, 757 received PROCRIT® and 125 received placebo. Of the 757 patients who received PROCRIT®, 361 (47%) were aged 65 and over, while 100 (13%) were 75 and over. No differences in safety or effectiveness were observed between geriatric and younger patients. Dose selection and adjustment for an elderly patient should be individualized to achieve and maintain the target hematocrit (See DOSAGE AND ADMINISTRATION ).
Insufficient numbers of patients age 65 or older were enrolled in clinical studies of PROCRIT® for the treatment of anemia associated with pre-dialysis chronic renal failure, cancer chemotherapy, and Zidovudine-treatment of HIV infection to determine whether they respond differently from younger subjects.
Chronic Renal Failure Patients
Patients with CRF Not Requiring Dialysis
Blood pressure and hemoglobin should be monitored no less frequently than for patients maintained on dialysis. Renal function and fluid and electrolyte balance should be closely monitored, as an improved sense of well-being may obscure the need to initiate dialysis in some patients.
Hematology
Sufficient time should be allowed to determine a patient's responsiveness to a dosage of PROCRIT® before adjusting the dose. Because of the time required for erythropoiesis and the red cell half-life, an interval of 2 to 6 weeks may occur between the time of a dose adjustment (initiation, increase, decrease, or discontinuation) and a significant change in hemoglobin.
In order to avoid reaching the suggested target hemoglobin too rapidly, or exceeding the suggested target range (hemoglobin of 10 g/dL to 12 g/dL), the guidelines for dose and frequency of dose adjustments (see DOSAGE AND ADMINISTRATION ) should be followed.
For patients who respond to PROCRIT® with a rapid increase in hemoglobin (eg, more than 1 g/dL in any 2-week period), the dose of PROCRIT® should be reduced because of the possible association of excessive rate of rise of hemoglobin with an exacerbation of hypertension.
The elevated bleeding time characteristic of CRF decreases toward normal after correction of anemia in adult patients treated with PROCRIT®. Reduction of bleeding time also occurs after correction of anemia by transfusion.
Laboratory Monitoring
The hemoglobin should be determined twice a week until it has stabilized in the suggested target range and the maintenance dose has been established. After any dose adjustment, the hemoglobin should also be determined twice weekly for at least 2 to 6 weeks until it has been determined that the hemoglobin has stabilized in response to the dose change. The hemoglobin should then be monitored at regular intervals.
A complete blood count with differential and platelet count should be performed regularly. During clinical trials, modest increases were seen in platelets and white blood cell counts. While these changes were statistically significant, they were not clinically significant and the values remained within normal ranges.
In patients with CRF, serum chemistry values (including blood urea nitrogen [BUN], uric acid, creatinine, phosphorus, and potassium) should be monitored regularly. During clinical trials in adult patients on dialysis, modest increases were seen in BUN, creatinine, phosphorus, and potassium. In some adult patients with CRF not on dialysis treated with PROCRIT®, modest increases in serum uric acid and phosphorus were observed. While changes were statistically significant, the values remained within the ranges normally seen in patients with CRF.
Diet
As the hemoglobin increases and patients experience an improved sense of well-being and quality of life, the importance of compliance with dietary and dialysis prescriptions should be reinforced. In particular, hyperkalemia is not uncommon in patients with CRF. In US studies in patients on dialysis, hyperkalemia has occurred at an annualized rate of approximately 0.11 episodes per patient-year of PROCRIT® therapy, often in association with poor compliance to medication, diet, and/or dialysis.
Dialysis Management
Therapy with PROCRIT® results in an increase in hematocrit and a decrease in plasma volume which could affect dialysis efficiency. In studies to date, the resulting increase in hematocrit did not appear to adversely affect dialyzer function 9,10 or the efficiency of high flux hemodialysis. 11 During hemodialysis, patients treated with PROCRIT® may require increased anticoagulation with heparin to prevent clotting of the artificial kidney.
Patients who are marginally dialyzed may require adjustments in their dialysis prescription. As with all patients on dialysis, the serum chemistry values (including BUN, creatinine, phosphorus, and potassium) in patients treated with PROCRIT® should be monitored regularly to assure the adequacy of the dialysis prescription.
Information for Patients
In those situations in which the physician determines that a home dialysis patient can safely and effectively self-administer PROCRIT®, the patient should be instructed as to the proper dosage and administration. Home dialysis patients should be referred to the full " Information For Home Dialysis Patients " insert; it is not a disclosure of all possible effects. Patients should be informed of the signs and symptoms of allergic drug reaction and advised of appropriate actions. If home use is prescribed for a home dialysis patient, the patient should be thoroughly instructed in the importance of proper disposal and cautioned against the reuse of needles, syringes, or drug product. A puncture-resistant container for the disposal of used syringes and needles should be available to the patient. The full container should be disposed of according to the directions provided by the physician.
Renal Function
In adult patients with CRF not on dialysis, renal function and fluid and electrolyte balance should be closely monitored, as an improved sense of well-being may obscure the need to initiate dialysis in some patients. In patients with CRF not on dialysis, placebo-controlled studies of progression of renal dysfunction over periods of greater than 1 year have not been completed. In shorter term trials in adult patients with CRF not on dialysis, changes in creatinine and creatinine clearance were not significantly different in patients treated with PROCRIT® compared with placebo-treated patients. Analysis of the slope of 1/serum creatinine versus time plots in these patients indicates no significant change in the slope after the initiation of PROCRIT® therapy.
Zidovudine-treated HIV-infected Patients
Hypertension
Exacerbation of hypertension has not been observed in zidovudine-treated HIV-infected patients treated with PROCRIT®. However, PROCRIT® should be withheld in these patients if pre-existing hypertension is uncontrolled, and should not be started until blood pressure is controlled. In double-blind studies, a single seizure has been experienced by a patient treated with PROCRIT®. 25
Cancer Patients on Chemotherapy
Hypertension
Hypertension, associated with a significant increase in hemoglobin, has been noted rarely in patients treated with PROCRIT®. Nevertheless, blood pressure in patients treated with PROCRIT® should be monitored carefully, particularly in patients with an underlying history of hypertension or cardiovascular disease.
Seizures
In double-blind, placebo-controlled trials, 3.2% (n = 2/63) of patients treated with PROCRIT® TIW and 2.9% (n = 2/68) of placebo-treated patients had seizures. Seizures in 1.6% (n = 1/63) of patients treated with PROCRIT® TIW occurred in the context of a significant increase in blood pressure and hematocrit from baseline values. However, both patients treated with PROCRIT® also had underlying CNS pathology which may have been related to seizure activity.
In a placebo-controlled, double-blind trial utilizing weekly dosing with PROCRIT®, 1.2% (n = 2/168) of safety-evaluable patients treated with PROCRIT® and 1% (n = 1/165) of placebo-treated patients had seizures. Seizures in the patients treated with weekly PROCRIT® occurred in the context of a significant increase in hemoglobin from baseline values, however, significant increases in blood pressure were not seen. These patients may have had other CNS pathology.
Thrombotic Events
In double-blind, placebo-controlled trials, 3.2% (n = 2/63) of patients treated with PROCRIT® TIW and 11.8% (n = 8/68) of placebo-treated patients had thrombotic events (eg, pulmonary embolism, cerebrovascular accident) (See WARNINGS ; Thrombotic Events and Increased Mortality ).
In a placebo-controlled, double-blind trial utilizing weekly dosing with PROCRIT®, 6.0% (n = 10/168) of safety-evaluable patients treated with PROCRIT® and 3.6% (n = 6/165) (p = 0.444) of placebo-treated patients had clinically significant thrombotic events (deep vein thrombosis requiring anticoagulant therapy, embolic event including pulmonary embolism, myocardial infarction, cerebral ischemia, left ventricular failure and thrombotic microangiopathy). A definitive relationship between the rate of hemoglobin increase and the occurrence of clinically significant thrombotic events could not be evaluated due to the limited schedule of hemoglobin measurements in this study.
Tumor Growth Factor Potential
PROCRIT® is a growth factor that primarily stimulates red cell production. Erythropoietin receptors are also found to be present on the surface of some malignant cell lines and tumor biopsy specimens. However, it is not known if these receptors are functional. A randomized, placebo-controlled trial was conducted in 224 chemotherapy-naive, non-anemic patients with small cell lung cancer receiving cisplatin-based combination chemotherapy, to investigate whether the concurrent use of PROCRIT® stimulated tumor growth as assessed by impact on overall response rate. Patients were randomized to receive PROCRIT® 150 Units/kg or placebo subcutaneously TIW during chemotherapy. The overall response rates, after 3 cycles of treatment, were 72% and 67%, in the PROCRIT® and placebo arms, respectively. Complete response rates (17% vs. 14%) and median overall survival (10.5 mos vs. 10.4 mos) were similar in the PROCRIT® and placebo arms. 25
Two additional studies explored effect on survival and/or progression of administrations of other exogenous erythropoietin with higher hemoglobin targets.
In a randomized, placebo-controlled study using another Epoetin alfa product, conducted in 939 women with metastatic breast cancer, study drug dosing was titrated to attempt to maintain hemoglobin levels between 12 and 14 g/dL. At four months, death attributed to disease progression was higher (6% vs 3%) in women receiving Epoetin alfa. Overall mortality was significantly higher at 12 months in the Epoetin alfa arm (See WARNINGS ; Thrombotic Events and Increased Mortality ).
In a randomized, placebo-controlled study using Epoetin beta, conducted in 351 patients with head and neck cancer, study drug was administered with the aim of achieving a hemoglobin level of 14 g/dL in women and 15 g/dL in men. Locoregional progression-free survival was significantly shorter (median PFS: 406 days Epoetin beta vs 745 days placebo, p = 0.04) in patients receiving Epoetin beta. 43
There is insufficient information to establish whether use of Epoetin products, including PROCRIT®, have an adverse effect on time to tumor progression or progression-free survival.
These trials permitted or required dosing to achieve hemoglobin of greater than 12 g/dL. Until further information is available, the recommended target hemoglobin should not exceed 12 g/dL in men or women.
Surgery Patients
Thrombotic/Vascular Events
In perioperative clinical trials with orthopedic patients, the overall incidence of thrombotic/vascular events was similar in Epoetin alfa and placebo-treated patients who had a pretreatment hemoglobin of > 10 g/dL to </= 13 g/dL. In patients with a hemoglobin of > 13 g/dL treated with 300 Units/kg of Epoetin alfa, the possibility that PROCRIT® treatment may be associated with an increased risk of postoperative thrombotic/vascular events cannot be excluded. 18-20,28
In one study in which Epoetin alfa was administered in the perioperative period to patients undergoing coronary artery bypass graft surgery, there were 7 deaths in the group treated with Epoetin alfa (n = 126) and no deaths in the placebo-treated group (n = 56). Among the 7 deaths in the patients treated with Epoetin alfa, 4 were at the time of therapy (between study day 2 and 8). The 4 deaths at the time of therapy (3%) were associated with thrombotic/vascular events. A causative role of Epoetin alfa cannot be excluded (see WARNINGS ).
Hypertension
Blood pressure may rise in the perioperative period in patients being treated with PROCRIT®. Therefore, blood pressure should be monitored carefully.
ADVERSE REACTIONS
Immunogenicity
As with all therapeutic proteins, there is the potential for immunogenicity. The observed incidence of antibody positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to PROCRIT® with the incidence of antibodies to other products may be misleading.
A few cases of PRCA associated with antibodies with neutralizing activity have been reported in patients receiving PROCRIT® (see WARNINGS : PURE RED CELL APLASIA ). These cases were observed in patients treated by either SC or IV routes of administration and occurred predominantly in CRF patients.
Chronic Renal Failure Patients
PROCRIT® is generally well-tolerated. The adverse events reported are frequent sequelae of CRF and are not necessarily attributable to PROCRIT® therapy. In double-blind, placebo-controlled studies involving over 300 patients with CRF, the events reported in greater than 5% of patients treated with PROCRIT® during the blinded phase were:
EventPercent of Patients Reporting Event Patients Treated
With PROCRIT®
(n = 200)Placebo-treated
Patients
(n = 135)Hypertension24% 19% Headache16% 12% Arthralgias11% 6% Nausea11% 9% Edema9% 10% Fatigue9% 14% Diarrhea9% 6% Vomiting8% 5% Chest Pain7% 9% Skin Reaction7% 12% (Administration Site)Asthenia7% 12% Dizziness7% 13% Clotted Access7% 2% Significant adverse events of concern in patients with CRF treated in double-blind, placebo-controlled trials occurred in the following percent of patients during the blinded phase of the studies:
Seizure1.1% 1.1% CVA/TIA0.4% 0.6% MI0.4% 1.1% Death0 1.7% In the US PROCRIT® studies in adult patients on dialysis (over 567 patients), the incidence (number of events per patient-year) of the most frequently reported adverse events were: hypertension (0.75), headache (0.40), tachycardia (0.31), nausea/vomiting (0.26), clotted vascular access (0.25), shortness of breath (0.14), hyperkalemia (0.11), and diarrhea (0.11). Other reported events occurred at a rate of less than 0.10 events per patient per year.
Events reported to have occurred within several hours of administration of PROCRIT® were rare, mild, and transient, and included injection site stinging in dialysis patients and flu-like symptoms such as arthralgias and myalgias.
In all studies analyzed to date, PROCRIT® administration was generally well-tolerated, irrespective of the route of administration.
Pediatric CRF Patients: In pediatric patients with CRF on dialysis, the pattern of most adverse events was similar to that found in adults. Additional adverse events reported during the double-blind phase in > 10% of pediatric patients in either treatment group were: abdominal pain, dialysis access complications including access infections and peritonitis in those receiving peritoneal dialysis, fever, upper respiratory infection, cough, pharyngitis, and constipation. The rates are similar between the treatment groups for each event.
Hypertension: Increases in blood pressure have been reported in clinical trials, often during the first 90 days of therapy. On occasion, hypertensive encephalopathy and seizures have been observed in patients with CRF treated with PROCRIT®. When data from all patients in the US phase 3 multicenter trial were analyzed, there was an apparent trend of more reports of hypertensive adverse events in patients on dialysis with a faster rate of rise of hematocrit (greater than 4 hematocrit points in any 2-week period). However, in a double-blind, placebo-controlled trial, hypertensive adverse events were not reported at an increased rate in the group treated with PROCRIT® (150 Units/kg TIW) relative to the placebo group.
Seizures: There have been 47 seizures in 1010 patients on dialysis treated with PROCRIT® in clinical trials, with an exposure of 986 patient-years for a rate of approximately 0.048 events per patient-year. However, there appeared to be a higher rate of seizures during the first 90 days of therapy (occurring in approximately 2.5% of patients) when compared to subsequent 90-day periods. The baseline incidence of seizures in the untreated dialysis population is difficult to determine; it appears to be in the range of 5% to 10% per patient-year. 39-41
Thrombotic Events: In clinical trials where the maintenance hematocrit was 35 ± 3% on PROCRIT®, clotting of the vascular access (A-V shunt) has occurred at an annualized rate of about 0.25 events per patient-year, and other thrombotic events (eg, myocardial infarction, cerebral vascular accident, transient ischemic attack, and pulmonary embolism) occurred at a rate of 0.04 events per patient-year. In a separate study of 1111 untreated dialysis patients, clotting of the vascular access occurred at a rate of 0.50 events per patient-year. However, in CRF patients on hemodialysis who also had clinically evident ischemic heart disease or congestive heart failure, the risk of A-V shunt thrombosis was higher (39% vs 29%, p < 0.001), and myocardial infarctions, vascular ischemic events, and venous thrombosis were increased, in patients targeted to a hematocrit of 42 ± 3% compared to those maintained at 30 ± 3% (see WARNINGS ).
In patients treated with commercial PROCRIT®, there have been rare reports of serious or unusual thrombo-embolic events including migratory thrombophlebitis, microvascular thrombosis, pulmonary embolus, and thrombosis of the retinal artery, and temporal and renal veins. A causal relationship has not been established.
Allergic Reactions: There have been no reports of serious allergic reactions or anaphylaxis associated with PROCRIT® administration during clinical trials. Skin rashes and urticaria have been observed rarely and when reported have generally been mild and transient in nature.
There have been rare reports of potentially serious allergic reactions including urticaria with associated respiratory symptoms or circumoral edema, or urticaria alone. Most reactions occurred in situations where a causal relationship could not be established. Symptoms recurred with rechallenge in a few instances, suggesting that allergic reactivity may occasionally be associated with PROCRIT® therapy. If an anaphylactoid reaction occurs, PROCRIT® should be immediately discontinued and appropriate therapy initiated.
Zidovudine-treated HIV-infected Patients
Adverse events reported in clinical trials with PROCRIT® in zidovudine-treated HIV-infected patients were consistent with the progression of HIV infection. In double-blind, placebo-controlled studies of 3 months duration involving approximately 300 zidovudine-treated HIV-infected patients, adverse events with an incidence of >/= 10% in either patients treated with PROCRIT® or placebo-treated patients were:
EventPercent of Patients Reporting Event Patients Treated
With PROCRIT®
(n = 144)Placebo-treated
Patients
(n = 153)Pyrexia38% 29% Fatigue25% 31% Headache19% 14% Cough18% 14% Diarrhea16% 18% Rash16% 8% Congestion,
Respiratory15% 10% Nausea15% 12% Shortness of Breath14% 13% Asthenia11% 14% Skin Reaction,(Medication Site)10% 7% Dizziness9% 10% In the 297 patients studied, PROCRIT® was not associated with significant increases in opportunistic infections or mortality. 25 In 71 patients from this group treated with PROCRIT® at 150 Units/kg TIW, serum p24 antigen levels did not appear to increase. 27 Preliminary data showed no enhancement of HIV replication in infected cell lines in vitro . 25
Peripheral white blood cell and platelet counts are unchanged following PROCRIT® therapy.
Allergic Reactions: Two zidovudine-treated HIV-infected patients had urticarial reactions within 48 hours of their first exposure to study medication. One patient was treated with PROCRIT® and one was treated with placebo (PROCRIT® vehicle alone). Both patients had positive immediate skin tests against their study medication with a negative saline control. The basis for this apparent pre-existing hypersensitivity to components of the PROCRIT® formulation is unknown, but may be related to HIV-induced immunosuppression or prior exposure to blood products.
Seizures: In double-blind and open-label trials of PROCRIT® in zidovudine-treated HIV-infected patients, 10 patients have experienced seizures. 25 In general, these seizures appear to be related to underlying pathology such as meningitis or cerebral neoplasms, not PROCRIT® therapy.
Cancer Patients on Chemotherapy
Adverse experiences reported in clinical trials with PROCRIT® administered TIW in cancer patients were consistent with the underlying disease state. In double-blind, placebo-controlled studies of up to 3 months duration involving 131 cancer patients, adverse events with an incidence > 10% in either patients treated with PROCRIT® or placebo-treated patients were as indicated below:
EventPercent of Patients Reporting Event Patients Treated
With PROCRIT®
(n = 63)Placebo-treated
Patients
(n = 68)Pyrexia29% 19% Diarrhea21% * 7% Nausea17% * 32% Vomiting17% 15% Edema17% * 1% Asthenia13% 16% Fatigue13% 15% Shortness of Breath13% 9% Paresthesia11% 6% Upper Respiratory Infection11% 4% Dizziness5% 12% Trunk Pain3% * 16% * Statistically significant Although some statistically significant differences between patients being treated with PROCRIT® and placebo-treated patients were noted, the overall safety profile of PROCRIT® appeared to be consistent with the disease process of advanced cancer. During double-blind and subsequent open-label therapy in which patients (n = 72 for total exposure to PROCRIT®) were treated for up to 32 weeks with doses as high as 927 Units/kg, the adverse experience profile of PROCRIT® was consistent with the progression of advanced cancer.
Three hundred thirty-three (333) cancer patients enrolled in a placebo-controlled, double-blind trial utilizing weekly dosing with PROCRIT® for up to 4 months were evaluable for adverse events. The incidence of adverse events was similar in both treatment and placebo arms.
Surgery Patients
Adverse events with an incidence of >/= 10% are shown in the following table:
Event
Percent of Patients Reporting Event Patients
Treated With PROCRIT®
300 U/kg
(n = 112) aPatients
Treated With PROCRIT®
100 U/kg
(n = 101) aPlacebo-
treated
Patients(n = 103) a
Patients
Treated With PROCRIT®
600 U/kg
(n = 73) bPatients
Treated With PROCRIT®
300 U/kg
(n = 72) bPyrexia51% 50% 60% 47% 42% Nausea48% 43% 45% 45% 58% Constipation43% 42% 43% 51% 53% Skin Reaction
(Medication Site)25% 19% 22% 26% 29% Vomiting22% 12% 14% 21% 29% Skin Pain18% 18% 17% 5% 4% Pruritus16% 16% 14% 14% 22% Insomnia13% 16% 13% 21% 18% Headache13% 11% 9% 10% 19% Dizziness12% 9% 12% 11% 21% Urinary Tract
Infection12% 3% 11% 11% 8% Hypertension10% 11% 10% 5% 10% Diarrhea10% 7% 12% 10% 6% Deep Venous
Thrombosis10% 3% 5% 0% 0% Dyspepsia9% 11% 6% 7% 8% Anxiety7% 2% 11% 11% 4% Edema6% 11% 8% 11% 7% a Study including patients undergoing orthopedic surgery treated with PROCRIT® or placebo for 15 days b Study including patients undergoing orthopedic surgery treated with PROCRIT® 600 Units/kg weekly × 4 or 300 Units/kg daily × 15 c Determined by clinical symptoms Thrombotic/Vascular Events: In three double-blind, placebo-controlled orthopedic surgery studies, the rate of deep venous thrombosis (DVT) was similar among Epoetin alfa and placebo-treated patients in the recommended population of patients with a pretreatment hemoglobin of > 10 g/dL to </= 13 g/dL. 18,20,28 However, in 2 of 3 orthopedic surgery studies the overall rate (all pretreatment hemoglobin groups combined) of DVTs detected by postoperative ultrasonography and/or surveillance venography was higher in the group treated with Epoetin alfa than in the placebo-treated group (11% vs 6%). This finding was attributable to the difference in DVT rates observed in the subgroup of patients with pretreatment hemoglobin > 13 g/dL. However, the incidence of DVTs was within the range of that reported in the literature for orthopedic surgery patients.
In the orthopedic surgery study of patients with pretreatment hemoglobin of > 10 g/dL to </= 13 g/dL which compared two dosing regimens (600 Units/kg weekly × 4 and 300 Units/kg daily × 15), 4 subjects in the 600 Units/kg weekly PROCRIT® group (5%) and no subjects in the 300 Units/kg daily group had a thrombotic vascular event during the study period. 19
In a study examining the use of Epoetin alfa in 182 patients scheduled for coronary artery bypass graft surgery, 23% of patients treated with Epoetin alfa and 29% treated with placebo experienced thrombotic/vascular events. There were 4 deaths among the Epoetin alfa-treated patients that were associated with a thrombotic/vascular event. A causative role of Epoetin alfa cannot be excluded (see WARNINGS ).
OVERDOSAGE
The maximum amount of PROCRIT® that can be safely administered in single or multiple doses has not been determined. Doses of up to 1500 Units/kg TIW for 3 to 4 weeks have been administered to adults without any direct toxic effects of PROCRIT® itself. 6 Therapy with PROCRIT® can result in polycythemia if the hemoglobin is not carefully monitored and the dose appropriately adjusted. If the suggested target range is exceeded, PROCRIT® may be temporarily withheld until the hemoglobin returns to the suggested target range; PROCRIT® therapy may then be resumed using a lower dose (see DOSAGE AND ADMINISTRATION ). If polycythemia is of concern, phlebotomy may be indicated to decrease the hemoglobin.
DOSAGE AND ADMINISTRATION
Chronic Renal Failure Patients
The recommended range for the starting dose of PROCRIT® is 50 to 100 Units/kg TIW for adult patients. The recommended starting dose for pediatric CRF patients on dialysis is 50 Units/kg TIW. The dose of PROCRIT® should be reduced as the hemoglobin approaches 12 g/dL or increases by more than 1 g/dL in any 2-week period. The dosage of PROCRIT® must be individualized to maintain the hemoglobin within the suggested target range. At the physician's discretion, the suggested target hemoglobin range may be expanded to achieve maximal patient benefit.
PROCRIT® may be given either as an IV or SC injection. In patients on hemodialysis, PROCRIT® usually has been administered as an IV bolus TIW. While the administration of PROCRIT® is independent of the dialysis procedure, PROCRIT® may be administered into the venous line at the end of the dialysis procedure to obviate the need for additional venous access. In adult patients with CRF not on dialysis, PROCRIT® may be given either as an IV or SC injection.
Patients who have been judged competent by their physicians to self-administer PROCRIT® without medical or other supervision may give themselves either an IV or SC injection. The table below provides general therapeutic guidelines for patients with CRF:
Starting Dose:Adults50 to 100 Units/kg TIW; IV or SCPediatric Patients50 Units/kg TIW; IV or SCReduce Dose When:1. Hgb approaches 12 g/dL or,2. Hgb increases > 1 g/dL in any 2-week periodIncrease Dose If:Hgb does not increase by 2 g/dL after 8 weeks of therapy, and hgb is below suggested target rangeMaintenance Dose:Individually titrateSuggested Target Hgb Range:10 g/dL to 12 g/dLDuring therapy, hematological parameters should be monitored regularly (see LABORATORY MONITORING ).
Pretherapy Iron Evaluation: Prior to and during PROCRIT® therapy, the patient's iron stores, including transferrin saturation (serum iron divided by iron binding capacity) and serum ferritin, should be evaluated. Transferrin saturation should be at least 20%, and ferritin should be at least 100 ng/mL. Virtually all patients will eventually require supplemental iron to increase or maintain transferrin saturation to levels that will adequately support erythropoiesis stimulated by PROCRIT®.
Dose Adjustment: The dose should be adjusted for each patient to achieve and maintain a target hemoglobin not to exceed 12 g/dL.
Increases in dose should not be made more frequently than once a month. If the hemoglobin is increasing and approaching 12 g/dL, the dose should be reduced by approximately 25%. If the hemoglobin continues to increase, dose should be temporarily withheld until the hemoglobin begins to decrease, at which point therapy should be reinitiated at a dose approximately 25% below the previous dose. If the hemoglobin increases by more than 1 g/dL in a 2-week period, the dose should be decreased by approximately 25%.
If the increase in the hemoglobin is less than 1 g/dL over 4 weeks and iron stores are adequate (see PRECAUTIONS : LABORATORY MONITORING ), the dose of PROCRIT® may be increased by approximately 25% of the previous dose. Further increases may be made at 4-week intervals until the specified hemoglobin is obtained.
Maintenance Dose: The maintenance dose must be individualized for each patient on dialysis. In the US phase 3 multicenter trial in patients on hemodialysis, the median maintenance dose was 75 Units/kg TIW, with a range from 12.5 to 525 Units/kg TIW. Almost 10% of the patients required a dose of 25 Units/kg, or less, and approximately 10% of the patients required more than 200 Units/kg TIW to maintain their hematocrit in the suggested target range. In pediatric hemodialysis and peritoneal dialysis patients, the median maintenance dose was 167 Units/kg/week (49 to 447 Units/kg per week) and 76 Units/kg per week (24 to 323 Units/kg/week) administered in divided doses (TIW or BIW), respectively to achieve the target range of 30% to 36%.
If the hemoglobin remains below, or falls below, the suggested target range, iron stores should be re-evaluated. If the transferrin saturation is less than 20%, supplemental iron should be administered. If the transferrin saturation is greater than 20%, the dose of PROCRIT® may be increased. Such dose increases should not be made more frequently than once a month, unless clinically indicated, as the response time of the hemoglobin to a dose increase can be 2 to 6 weeks. Hemoglobin should be measured twice weekly for 2 to 6 weeks following dose increases. In adult patients with CRF not on dialysis, the maintenance dose must also be individualized. PROCRIT® doses of 75 to 150 Units/kg/week have been shown to maintain hematocrits of 36% to 38% for up to 6 months.
Lack or Loss of Response: Over 95% of patients with CRF responded with clinically significant increases in hematocrit, and virtually all patients were transfusion-independent within approximately 2 months of initiation of PROCRIT® therapy.
If a patient fails to respond or maintain a response, other etiologies should be considered and evaluated as clinically indicated (see PRECAUTIONS : LACK OR LOSS OF RESPONSE ).
Zidovudine-treated HIV-infected Patients
Prior to beginning PROCRIT®, it is recommended that the endogenous serum erythropoietin level be determined (prior to transfusion). Available evidence suggests that patients receiving zidovudine with endogenous serum erythropoietin levels > 500 mUnits/mL are unlikely to respond to therapy with PROCRIT®.
Starting Dose: For adult patients with serum erythropoietin levels </= 500 mUnits/mL who are receiving a dose of zidovudine </= 4200 mg/week, the recommended starting dose of PROCRIT® is 100 Units/kg as an IV or SC injection TIW for 8 weeks. For pediatric patients, see PRECAUTIONS : PEDIATRIC USE .
Increase Dose: During the dose adjustment phase of therapy, the hemoglobin should be monitored weekly. If the response is not satisfactory in terms of reducing transfusion requirements or increasing hemoglobin after 8 weeks of therapy, the dose of PROCRIT® can be increased by 50 to 100 Units/kg TIW. Response should be evaluated every 4 to 8 weeks thereafter and the dose adjusted accordingly by 50 to 100 Units/kg increments TIW. If patients have not responded satisfactorily to a PROCRIT® dose of 300 Units/kg TIW, it is unlikely that they will respond to higher doses of PROCRIT®.
Maintenance Dose: After attainment of the desired response (ie, reduced transfusion requirements or increased hemoglobin), the dose of PROCRIT® should be titrated to maintain the response based on factors such as variations in zidovudine dose and the presence of intercurrent infectious or inflammatory episodes. If the hemoglobin exceeds 13 g/dL, the dose should be discontinued until the hemoglobin drops to 12 g/dL. The dose should be reduced by 25% when treatment is resumed and then titrated to maintain the desired hemoglobin.
Cancer Patients on Chemotherapy
Although no specific serum erythropoietin level can be stipulated above which patients would be unlikely to respond to PROCRIT® therapy, treatment of patients with grossly elevated serum erythropoietin levels (eg, > 200 mUnits/mL) is not recommended. The hemoglobin should be monitored on a weekly basis in patients receiving PROCRIT® therapy until hemoglobin becomes stable. The dose of PROCRIT® should be titrated to maintain the desired hemoglobin.
Two PROCRIT® dosing regimens may be used in adults; 150 Units/kg SC TIW or 40,000 Units SC Weekly. For pediatric patients, see PRECAUTIONS : PEDIATRIC USE .
TIW Dosing
Starting Dose:Adults150 Units/kg SC TIWPediatric PatientsSee PRECAUTIONS : Pediatric Use .Reduce Dose by 25% when:1. Hgb approaches 12 g/dL or,2. Hgb increases > 1 g/dL in any 2-week periodWithhold Dose if:Hgb exceeds 13 g/dL, until the hemoglobin fall to 12 g/dL, and restart dose at 25% below the previous doseIncrease Dose to
300 Units/kg TIW if:response is not satisfactory [no reduction in transfusion requirements or rise in hemoglobin] after 8 weeksSuggested Target Hgb Range:10 g/dL to 12 g/dLDuring therapy, hematological parameters should be monitored regularly (see PRECAUTIONS : Laboratory Monitoring ).
Weekly Dosing
- The starting dose in adults is 40,000 Units SC Weekly. If after 4 weeks of therapy, the hemoglobin has not increased by >/= 1 g/dL, in the absence of RBC transfusion, the PROCRIT® dose should be increased to 60,000 Units Weekly.
- If patients have not responded satisfactorily to a PROCRIT® dose of 60,000 Units Weekly after 4 weeks, it is unlikely that they will respond to higher doses of PROCRIT®.
- PROCRIT® should be withheld if the hemoglobin exceeds 13 g/dL and reinitiated with a 25% dose reduction when the hemoglobin is less than 12 g/dL.
- If PROCRIT® treatment produces a very rapid hemoglobin response (e.g., an increase of more than 1 g/dL in any 2-week period), the dose of PROCRIT® should be reduced by 25%.
Surgery Patients
Prior to initiating treatment with PROCRIT® a hemoglobin should be obtained to establish that it is > 10 to </= 13 g/dL. 18 The recommended dose of PROCRIT® is 300 Units/kg/day subcutaneously for 10 days before surgery, on the day of surgery, and for 4 days after surgery.
An alternate dose schedule is 600 Units/kg PROCRIT® subcutaneously in once weekly doses (21, 14, and 7 days before surgery) plus a fourth dose on the day of surgery. 19
All patients should receive adequate iron supplementation. Iron supplementation should be initiated no later than the beginning of treatment with PROCRIT® and should continue throughout the course of therapy.
PREPARATION AND ADMINISTRATION OF PROCRIT®
- Do not shake. It is not necessary to shake PROCRIT®. Prolonged vigorous shaking may denature any glycoprotein, rendering it biologically inactive.
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration. Do not use any vials exhibiting particulate matter or discoloration.
- Using aseptic techniques, attach a sterile needle to a sterile syringe. Remove the flip top from the vial containing PROCRIT®, and wipe the septum with a disinfectant. Insert the needle into the vial, and withdraw into the syringe an appropriate volume of solution.
-
Single-dose:
1 mL vial contains no preservative. Use one dose per vial; do not re-enter the vial. Discard unused portions.
Multidose: 1 mL and 2 mL vials contain preservative. Store at 2° to 8°C after initial entry and between doses. Discard 21 days after initial entry. - Do not dilute or administer in conjunction with other drug solutions. However, at the time of SC administration, preservative-free PROCRIT® from single-use vials may be admixed in a syringe with bacteriostatic 0.9% sodium chloride injection, USP, with benzyl alcohol 0.9% (bacteriostatic saline) at a 1:1 ratio using aseptic technique. The benzyl alcohol in the bacteriostatic saline acts as a local anesthetic which may ameliorate SC injection site discomfort. Admixing is not necessary when using the multidose vials of PROCRIT® containing benzyl alcohol.
HOW SUPPLIED
PROCRIT®, containing Epoetin alfa, is available in vials containing color coded labels and caps.
1 mL Single-Dose, Preservative-free Solution
Each dosage form is supplied in the following packages:
Cartons containing six (6) single-dose vials:
2000 Units/mL (NDC 59676-302-01) (Purple)
3000 Units/mL (NDC 59676-303-01) (Magenta)
4000 Units/mL (NDC 59676-304-01) (Green)
10,000 Units/mL (NDC 59676-310-01) (Red)
Cartons containing four (4) single-dose vials:
40,000 Units/mL (NDC 59676-340-01) (Orange)
Trays containing twenty-five (25) single-dose vials:
2000 Units/mL (NDC 59676-302-02) (Purple)
3000 Units/mL (NDC 59676-303-02) (Magenta)
4000 Units/mL (NDC 59676-304-02) (Green)
10,000 Units/mL (NDC 59676-310-02) (Red)
2 mL Multidose, Preserved Solution
Cartons containing six (6) multidose vials:
10,000 Units/mL (NDC 59676-312-01) (Blue)
1 mL Multidose, Preserved Solution
Cartons containing six (6) multidose vials:
20,000 Units/mL (NDC 59676-320-01) (Lime)
STORAGE
Store at 2° to 8° C (36° to 46° F). Do not freeze or shake.
REFERENCES
- Egrie JC, Strickland TW, Lane J, et al. Characterization and Biological Effects of Recombinant Human Erythropoietin. Immunobiol. 1986;72:213-224.
- Graber SE, Krantz SB. Erythropoietin and the Control of Red Cell Production. Ann Rev Med. 1978;29:51-66.
- Eschbach JW, Adamson JW. Anemia of End-Stage Renal Disease (ESRD). Kidney Intl. 1985;28:1-5.
- Eschbach JW, Egrie JC, Downing MR, et al. Correction of the Anemia of End-Stage Renal Disease with Recombinant Human Erythropoietin. NEJM. 1987;316:73-78.
- Eschbach JW, Abdulhadi MH, Browne JK, et al. Recombinant Human Erythropoietin in Anemic Patients with End-Stage Renal Disease. Ann Intern Med. 1989;111:992-1000.
- Eschbach JW, Egrie JC, Downing MR, et al. The Use of Recombinant Human Erythropoietin (r-HuEPO): Effect in End-Stage Renal Disease (ESRD). In: Friedman, Beyer, DeSanto, Giordano, eds. Prevention Of Chronic Uremia. Philadelphia, PA: Field and Wood Inc. 1989;148-155.
- Egrie JC, Eschbach JW, McGuire T, Adamson JW. Pharmacokinetics of Recombinant Human Erythropoietin (r-HuEPO) Administered to Hemodialysis (HD) Patients. Kidney Intl. 1988;33:262.
- Evans RW, Rader B, Manninen DL, et al. The Quality of Life of Hemodialysis Recipients Treated with Recombinant Human Erythropoietin. JAMA. 1990;263:825-830.
- Paganini E, Garcia J, Ellis P, et al. Clinical Sequelae of Correction of Anemia with Recombinant Human Erythropoietin (r-HuEPO); Urea Kinetics, Dialyzer Function and Reuse. Am J Kid Dis. 1988;11:16.
- Delano BG, Lundin AP, Golansky R, et al. Dialyzer Urea and Creatinine Clearances Not Significantly Changed in r-HuEPO Treated Maintenance Hemodialysis (MD) Patients. Kidney Intl. 1988;33:219.
- Stivelman J, Van Wyck D, and Ogden D. Use of Recombinant Erythropoietin (r-HuEPO) with High Flux Dialysis (HFD) Does Not Worsen Azotemia or Shorten Access Survival. Kidney Intl. 1988;33:239.
- Lim VS, DeGowin RL, Zavala D, et al. Recombinant Human Erythropoietin Treatment in Pre-Dialysis Patients: A Double-Blind Placebo Controlled Trial. Ann Int Med. 1989;110:108-114.
- Stone WJ, Graber SE, Krantz SB, et al. Treatment of the Anemia of Pre-Dialysis Patients with Recombinant Human Erythropoietin: A Randomized, Placebo-Controlled Trial. Am J Med Sci. 1988;296:171-179.
- Braun A, Ding R, Seidel C, Fies T, Kurtz A, Scharer K. Pharmacokinetics of recombinant human erythropoietin applied subcutaneously to children with chronic renal failure. Pediatr Nephrol 1993;7:61-64.
- Geva P, Sherwood JB. Pharmacokinetics of recombinant human erythropoietin (rHuEPO) in pediatric patients on chronic cycling peritoneal dialysis (CCPD). Blood. 1991;78 (Suppl 1):91a.
- Jabs K, Grant JR, Harmon W, et al. Pharmacokinetics of Epoetin alfa (rHuEPO) in pediatric hemodialysis (HD) patients. J Am Soc Nephrol. 1991;2:380.
- Kling PJ, Widness JA, Guillery EN, Veng-Pedersen P, Peters C, DeAlarcon PA. Pharmacokinetics and pharmacodynamics of erythropoietin during therapy in an infant with renal failure. J Pediatr. 1992;121:822-825.
- de Andrade JR and Jove M. Baseline Hemoglobin as a Predictor of Risk of Transfusion and Response to Epoetin alfa in Orthopedic Surgery Patients. Am. J. of Orthoped. 1996;25 (8): 533-542.
- Goldberg MA and McCutchen JW. A Safety and Efficacy Comparison Study of Two Dosing Regimens of Epoetin alfa in Patients Undergoing Major Orthopedic Surgery. Am. J. of Orthoped. 1996;25 (8): 544-552.
- Faris PM and Ritter MA. The Effects of Recombinant Human Erythropoietin on Perioperative Transfusion Requirements in Patients Having a Major Orthopedic Operation. J. Bone and Joint Surgery. 1996;78-A:62-72.
- Lundin AP, Akerman MJH, Chesler RM, et al. Exercise in Hemodialysis Patients after Treatment with Recombinant Human Erythropoietin. Nephron. 1991;58:315-319.
- Amgen Inc., data on file.
- Eschbach JW, Kelly MR, Haley NR, et al. Treatment of the Anemia of Progressive Renal Failure with Recombinant Human Erythropoietin. NEJM. 1989;321:158-163.
- The US Recombinant Human Erythropoietin Predialysis Study Group. Double-Blind, Placebo-Controlled Study of the Therapeutic Use of Recombinant Human Erythropoietin for Anemia Associated with Chronic Renal Failure in Predialysis Patients. Am J Kid Dis. 1991;18:50-59.
- Ortho Biologics, Inc., data on file.
- Danna RP, Rudnick SA, Abels RI. Erythropoietin Therapy for the Anemia Associated with AIDS and AIDS Therapy and Cancer. In: MB Garnick, ed. Erythropoietin in Clinical Applications -- An International Perspective. New York, NY:Marcel Dekker. 1990;301-324.
- Fischl M, Galpin JE, Levine JD, et al. Recombinant Human Erythropoietin for Patients with AIDS Treated with Zidovudine. NEJM. 1990;322:1488-1493.
- Laupacis A. Effectiveness of Perioperative Recombinant Human Erythropoietin in Elective Hip Replacement. Lancet. 1993;341:1228-1232.
- Kerr DN. Chronic Renal Failure. In: Beeson PB, McDermott W, Wyngaarden JB, eds, Cecil Textbook of Medicine. Philadelphia, PA: W.B. Saunders; 1979;1351-1367.
- Campos A, Garin EH. Therapy of renal anemia in children and adolescents with recombinant human erythropoietin (rHuEPO). Clin Pediatr (Phila). 1992;31:94-99.
- Montini G, Zacchello G, Baraldi E, et al. Benefits and risks of anemia correction with recombinant human erythropoietin in children maintained by hemodialysis. J Pediatr. 1990;117:556-560.
- Offner G, Hoyer PF, Latta K, Winkler L, Brodehl J, Scigalla P. One year's experience with recombinant erythropoietin in children undergoing continuous ambulatory or cycling peritoneal dialysis. Pediatr Nephrol. 1990;4:498-500.
- Muller-Wiefel DE, Scigalla P. Specific problems of renal anemia in childhood. Contrib Nephrol. 1988;66:71-84.
- Scharer K, Klare B, Dressel P, Gretz N. Treatment of renal anemia by subcutaneous erythropoietin in children with preterminal chronic renal failure. Acta Paediatr. 1993;82:953-958.
- Mueller BU, Jacobsen RN, Jarosinski P, et al. Erythropoietin for zidovudine-associated anemia in children with HIV infection. Pediatr AIDS and HIV Infect: Fetus to Adolesc. 1994;5:169-173.
- Zuccotti GV, Plebani A, Biasucci G, et al. Granulocyte-colony stimulating factor and erythropoietin therapy in children with human immunodeficiency virus infection. J Int Med Res. 1996;24:115-121.
- Beck MN, Beck D. Recombinant erythropoietin in acute chemotherapy-induced anemia of children with cancer. Med Pediatr Oncol. 1995;25:17-21.
- Bennetts G, Bertolone S, Bray G, Dinndorf P, Feusner J, Cairo M. Erythropoietin reduces volumes of red cell transfusions required in some subsets of children with acute lymphocytic leukemia. Blood. 1995;86:853a.
- Raskin NH, Fishman RA. Neurologic Disorders in Renal Failure (First of Two Parts). NEJM. 1976;294:143-148.
- Raskin NH and Fishman RA. Neurologic Disorders in Renal Failure (Second of Two Parts). NEJM. 1976;294:204-210.
- Messing RO, Simon RP. Seizures as a Manifestation of Systemic Disease. Neurologic Clinics. 1986;4:563-584.
- Besarab A, Bolton WK, Browne JK, et al. The effects of normal as compared with low hematocrit values in patients with cardiac disease who are receiving hemodialysis and epoetin. NEJM. 1998;339:584-90.
- Henke, M, Laszig, R, Rübe, C, et al. Erythropoietin to treat head and neck cancer patients with anaemia undergoing radiotherapy: randomized, double-blind, placebo-controlled trial. The Lancet. 2003;362:1255-1260.
- Widness JA, Veng-Pedersen P, Peters C, Pereira LM, Schmidt RL, Lowe SL. Erythropoietin Pharmacokinetics in Premature Infants: Developmental, Nonlinearity, and Treatment Effects. J Appl Physiol. 1996;80(1): 140-148.
This product's label may have been revised after this insert was used in production. For further product information and the current package insert, please visit www.procrit.com or call our Medical Information Group toll-free at (800) 325-7504, prompt #2.
Manufactured by:
Amgen Inc.
One Amgen Center Drive
Thousand Oaks, CA 91320-1799
Distributed by:
Ortho Biotech Products, L.P.
Raritan, New Jersey 08869-0670
ORTHO BIOTECH
Revised January 2005
© OBPLP 2000
638-10-979-4
Printed in U. S. A.
08PCT1753R13
PROCRIT®
EPOETIN ALFAINFORMATION FOR HOME DIALYSIS PATIENTS
What is PROCRIT® and how does it work?
PROCRIT® is a copy of human erythropoietin, a hormone produced primarily by healthy kidneys. PROCRIT® replaces the erythropoietin that the failed kidneys can no longer produce, and signals the bone marrow to make the oxygen-carrying red blood cells once again. PROCRIT® is produced in mammalian cells that have been genetically altered by the addition of a gene of the natural substance erythropoietin.
How should I take PROCRIT®?
In those situations where your doctor has determined that you, as a home dialysis patient, can self-administer PROCRIT®, you will receive instruction on how much PROCRIT® to use, how to inject it, how often you should inject it, and how you should dispose of the unused portions of each vial. You will be instructed to monitor your blood pressure carefully every day and to report any changes outside of the guidelines that your doctor has given you. When the number of red blood cells increases, your blood pressure can also increase, so your doctor may prescribe some new or additional blood pressure medication. Be sure to follow your doctor's orders. You may also be instructed to have certain laboratory tests, such as additional hematocrit or iron level measurements, done more frequently. You may be asked to report these tests to your doctor or dialysis center. Also, your doctor may prescribe additional iron for you to take. Be sure to comply with your doctor's orders. Continue to check your access, as your doctor or nurse has shown you, to make sure it is working. Be sure to let your healthcare professional know right away if there is a problem.
Allergy to PROCRIT®
Patients occasionally experience redness, swelling, or itching at the site of injection of PROCRIT®. This may indicate an allergy to the components of PROCRIT®, or it may indicate a local reaction. If you have a local reaction, consult your doctor. A potentially more serious reaction would be a generalized allergy to PROCRIT®, which could cause a rash over the whole body, shortness of breath, wheezing, reduction in blood pressure, fast pulse, or sweating. Severe cases of generalized allergy may be life-threatening. If you think you are having a generalized allergic reaction, stop taking PROCRIT® and notify a doctor or emergency medical personnel immediately.
How will I know if PROCRIT® is working?
The effectiveness of PROCRIT® is measured by the increase in hematocrit (the amount of red blood cells in the blood) that results from PROCRIT® therapy. The rise in hematocrit is not immediate. It usually takes about 2 to 6 weeks before the hematocrit starts to rise. The amount of time it takes, and the dose of PROCRIT® that is needed to make the hematocrit increase, varies from patient to patient.
What is the most important information I should know about PROCRIT® and CHRONIC RENAL FAILURE?
PROCRIT® has been prescribed for you by your doctor because you:
- Have anemia due to your kidney disease.
- Are able to dialyze at home.
- Have been determined to be able to administer PROCRIT® without direct medical or other supervision.
A lack of energy or feeling of tiredness is the major symptom of anemia. Additional symptoms include shortness of breath, chestpain, and feeling cold all the time. The reason for these symptoms is that there is a lack of red blood cells. Red blood cells carry oxygen, which is important for all of the body's functions. When there are fewer red blood cells, the body does not get all the oxygen it needs.
Kidneys remove toxins from the blood; they also measure the amount of oxygen in the blood. If there is not enough oxygen, the kidneys will produce a hormone called erythropoietin. Erythropoietin is released into the bloodstream and travels to the bone marrow where red blood cells are made. Erythropoietin signals the bone marrow to make more oxygen-carrying red bloodcells.
As the kidneys fail, they stop cleansing toxins from your blood. They also make less erythropoietin than they should. Therefore, the bone marrow does not receive a strong-enough signal to make the oxygen-carrying red blood cells. Fewer red blood cells are produced so the muscles, brain, and other parts of the body do not get the oxygen they need to function properly.
Most patients treated with PROCRIT® no longer need blood transfusions. However, certain medical conditions, or unexpected blood loss, may result in the need for a transfusion.
What do I need to know if I am giving myself PROCRIT® injections?
When you receive your PROCRIT® from the dialysis center, doctor's office or home dialysis supplier, always check to see that:
- The name PROCRIT® appears on the carton and vial label.
- You will be able to use PROCRIT® before the expiration date stamped on the package.
The PROCRIT® solution in the vial should always be clear and colorless. Do not use PROCRIT® if the contents of the vial appear discolored or cloudy, or if the vial appears to contain lumps, flakes, or particles. In addition, if the vial has been shaken vigorously, the solution may appear to be frothy and should not be used. Therefore, care should be taken not to shake the PROCRIT® vial vigorously before use. Unless you have been prescribed Multidose PROCRIT® (1 mL or 2 mL vials with a big "M"on the label, each containing a total of 20,000 Units of PROCRIT®), vials of PROCRIT® are for single use. Any unused portion of a vial should not be used. However, Multidose PROCRIT® may be used to inject multiple doses as prescribed by your doctor, and may be stored in the refrigerator (but not the freezing compartment) between doses for up to 21 days, and can be used for multiple doses. Follow your doctor's or dialysis center's instructions on what to do with the used vials.
How should I store PROCRIT®?
PROCRIT® should be stored in the refrigerator, but not in the freezing compartment. Do not let the vial freeze and do not leave it in direct sunlight. Do not use a vial of PROCRIT® that has been frozen or after the expiration date that is stamped on the label. If you have any questions about the safety of a vial of PROCRIT® that has been subjected to temperature extremes,be sure to check with your dialysis unit staff.
Always use the correct syringe.
Your doctor has instructed you on how to give yourself the correct dosage of PROCRIT®. This dosage will usually be measured in Units per milliliter or cc's. It is important to use a syringe that is marked in tenths of milliliters (for example, 0.2 mL or cc). Failure to use the proper syringe can lead to a mistake in dosage, and you may receive too much or too little PROCRIT®. Too little PROCRIT® may not be effective in increasing your hematocrit, and too much PROCRIT® may lead to a hematocrit that is too high. Only use disposable syringes and needles as they do not require sterilization; they should be used once and disposed of as instructed by your doctor.
IMPORTANT: TO HELP AVOID CONTAMINATION AND POSSIBLE INFECTION, FOLLOW THESE INSTRUCTIONS EXACTLY.
PREPARING THE DOSE
1. Wash your hands thoroughly with soap and water before preparing the medication.
2. Check the date on the PROCRIT® vial to be sure that the drug has not expired.
3. Remove the vial of PROCRIT® from the refrigerator and allow it to reach room temperature. Unless you are using a Multidose vial, each PROCRIT® vial is designed to be used only once. It is not necessary to shake PROCRIT®. Prolonged vigorous shaking may damage the product. Assemble the other supplies you will need for your injection.
4. Hemodialysis patients should wipe off the venous port of the hemodialysis tubing with an antiseptic swab. Peritoneal dialysis patients should cleanse the skin with an antiseptic swab where the injection is to be made.
5. Flip off the protective cap but do not remove the gray rubber stopper. Wipe the top of the gray rubber stopper with an antiseptic swab.
6. Using a syringe and needle designed for subcutaneous injection, draw air into the syringe by pulling back on the plunger. The amount of air should be equal to your PROCRIT® dose.
7. Carefully remove the needle cover. Put the needle through the gray rubber stopper of the PROCRIT® vial.
8. Push the plunger in to discharge air into the vial. The air injected into the vial will allow PROCRIT® to be easily withdrawn into the syringe.
9. Turn the vial and syringe upside down in one hand. Be sure the tip of the needle is in the PROCRIT® solution. Your other hand will be free to move the plunger. Draw back on the plunger slowly to draw the correct dose of PROCRIT® into the syringe.
10. Check for air bubbles. The air is harmless, but too large an air bubble will reduce the PROCRIT® dose. To remove air bubbles, gently tap the syringe to move the air bubbles to the top of the syringe, then use the plunger to push the solution and the air back into the vial. Then re-measure your correct dose of PROCRIT®.
11. Double check your dose. Remove the needle from the vial. Do not lay the syringe down or allow the needle to touch anything.
INJECTING THE DOSE
Patients on home hemodialysis using the intravenous injection route:
1. Insert the needle of the syringe into the previously cleansed venous port and inject the PROCRIT®.
2. Remove the syringe and dispose of the whole unit. Use the disposable syringe only once. Dispose of syringes and needles as directed by your doctor, by following these simple steps:
- Place all used needles and syringe sin a hard plastic container with a screw-on-cap, or a metal container with a plastic lid, such as a coffee can properly labeled as to content. If a metal container is used, cut a small hole in the plastic lid and tape the lid to the metal container. If a hard-plastic container is used, always screw the cap on tightly after each use. When the container is full, tape around the cap or lid, and dispose of according to your doctor's instructions.
- Do not use glass or clear plastic containers, or any container that will be recycled or returned to a store.
- Always store the container out of the reach of children.
- Please check with your doctor, nurse, or pharmacist for other suggestions. There may be special state and local laws that they will discuss with you.
Patients on homeperitoneal dialysis or home hemodialysis using the subcutaneous route:
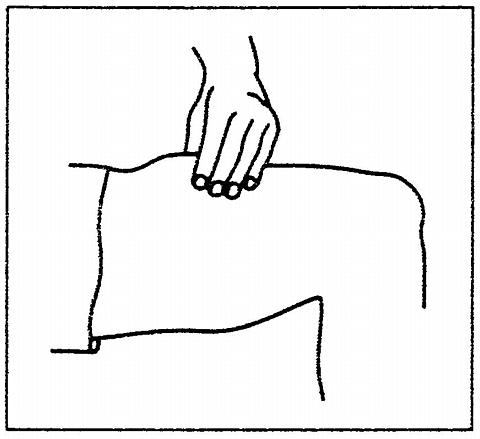
1. With one hand, stabilize the previously cleansed skin by spreading it or by pinching up a large area with your free hand.
2. Hold the syringe with the other hand, as you would a pencil. Double check that the correct amount of PROCRIT® is in the syringe. Insert the needle straight into the skin (90 degree angle). Pull the plunger back slightly. If blood comes into the syringe, do not inject PROCRIT®, as the needle has entered a blood vessel; withdraw the syringe and inject at a different site. Inject the PROCRIT® by pushing the plunger all the way down.
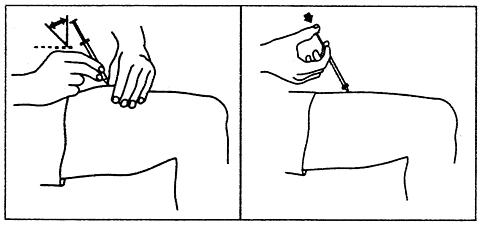
3. Hold an antiseptic swab near the needle and pull the needle straight out of the skin. Press the antiseptic swab over the injection site for several seconds.
4. Use the disposable syringe only once. Dispose of syringes and needles asdirected by your doctor, by following these simple steps:
- Place all used needles and syringes in a hard plastic container with a screw-on-cap, or a metal container with a plastic lid, such as a coffee can properly labeled as to content. If a metal container is used, cut a small hole in the plastic lid and tape the lid to the metal container. If a hard-plastic container is used, always screw the cap on tightly after each use. When the container is full, tape around the cap or lid, and dispose of according to your doctor's instructions.
- Do not use glass or clear plastic containers, or any container that will be recycled or returned to a store.
- Always store the container out of the reach of children.
- Please check with your doctor, nurse, or pharmacist for other suggestions. There may be special state and local laws that they will discuss with you.
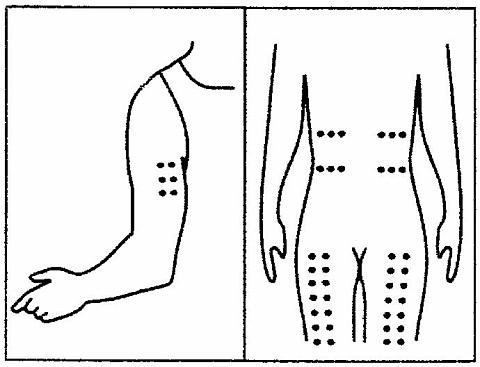
5. Always change the site for each injection as directed. Occasionally a problem may develop at the injection site. If you notice a lump, swelling, or bruising that doesn't go away, contact your doctor. You may wish to record the site just used so that you can keep track.
USAGE IN PREGNANCY
If you are pregnant or nursing a baby, consult your doctor before using PROCRIT®.
IMPORTANT NOTES
Since you are a home dialysis patient and your doctor allows you to self-administer PROCRIT®, please note the following:
- Always follow the instructions of your doctor concerning the dosage and administration of PROCRIT®. Do not change the dose or instructions for administration of PROCRIT® without consulting your doctor.
- Your doctor will tell you what to do if you miss a dose of PROCRIT®. Always keep a spare syringe and needle on hand.
- Always consult your doctor if you notice anything unusual about your condition or your use of PROCRIT®.
Manufactured by:
Amgen Inc.
One Amgen Center Drive
Thousand Oaks, CA 91320-1799
Distributed by:
Ortho Biotech Products, L.P.
Raritan, New Jersey 08869-0670
ORTHO BIOTECH
© OBPLP 2000
Printed in U.S.A.
Revised January 2005 638-10-979-4
08PCT1753R13
Subscribe to the "News" RSS Feed 
TOP ۞