-
Raptiva for Injection (Genentech)
DESCRIPTION
RAPTIVA® [efalizumab] is an immunosuppressive recombinant humanized IgG1 kappa isotype monoclonal antibody that binds to human CD11a ( 1 ). Efalizumab has a molecular weight of approximately 150 kilodaltons and is produced in a Chinese hamster ovary mammalian cell expression system in a nutrient medium containing the antibiotic gentamicin. Gentamicin is not detectable in the final product.
RAPTIVA is supplied as a sterile, white to off-white, lyophilized powder in single-use glass vials for subcutaneous (SC) injection. Reconstitution of the single-use vial with 1.3 mL of the supplied sterile water for injection (non-USP) yields approximately 1.5 mL of solution to deliver 125 mg per 1.25 mL (100 mg/mL) of RAPTIVA. The sterile water for injection supplied does not comply with USP requirement for pH. After reconstitution, RAPTIVA is a clear to pale yellow solution with a pH of approximately 6.2. Each single-use vial of RAPTIVA contains 150 mg of efalizumab, 123.2 mg of sucrose, 6.8 mg of L-histidine hydrochloride monohydrate, 4.3 mg of L-histidine and 3 mg of polysorbate 20 and is designed to deliver 125 mg of efalizumab in 1.25 mL.
CLINICAL PHARMACOLOGY
Mechanism of Action
RAPTIVA binds to CD11a, the (alpha) subunit of leukocyte function antigen-1 (LFA-1), which is expressed on all leukocytes, and decreases cell surface expression of CD11a. RAPTIVA inhibits the binding of LFA-1 to intercellular adhesion molecule-1 (ICAM-1), thereby inhibiting the adhesion of leukocytes to other cell types. Interaction between LFA-1 and ICAM-1 contributes to the initiation and maintenance of multiple processes, including activation of T lymphocytes, adhesion of T lymphocytes to endothelial cells, and migration of T lymphocytes to sites of inflammation including psoriatic skin. Lymphocyte activation and trafficking to skin play a role in the pathophysiology of chronic plaque psoriasis. In psoriatic skin, ICAM-1 cell surface expression is upregulated on endothelium and keratinocytes. CD11a is also expressed on the surface of B lymphocytes, monocytes, neutrophils, natural killer cells, and other leukocytes. Therefore, the potential exists for RAPTIVA to affect the activation, adhesion, migration, and numbers of cells other than T lymphocytes.
Pharmacokinetics
In patients with moderate to severe plaque psoriasis, following an initial SC RAPTIVA dose of 0.7 mg/kg followed by 11 weekly SC doses of 1 mg/kg/wk, serum concentrations reached a steady-state at 4 weeks with a mean trough concentration of approximately 9 µg/mL (n = 26). After the last dose, the mean peak concentration was approximately 12 µg/mL (n = 25). Mean steady-state clearance was 24 mL/kg/day (range = 5-76 mL/kg/day, n = 25). Mean time to eliminate RAPTIVA after the last steady-state dose was 25 days (range = 13-35 days, n = 17). The mean estimated RAPTIVA SC bioavailability was 50%. In a population pharmacokinetic analysis of 1088 patients, body weight was found to be the most significant covariate affecting RAPTIVA clearance. In patients receiving weekly SC doses of 1 mg/kg, RAPTIVA exposure was similar across body weight quartiles. RAPTIVA clearance was not significantly affected by gender or race. The pharmacokinetics of RAPTIVA in pediatric patients have not been studied. The effects of renal or hepatic impairment on the pharmacokinetics of RAPTIVA have not been studied.
Pharmacodynamics
At a dose of 1 mg/kg/wk SC, RAPTIVA reduced expression of CD11a on circulating T lymphocytes to approximately 15-25% of pre-dose values and reduced free CD11a binding sites to a mean of </=5% of pre-dose values. These pharmacodynamic effects were seen 1-2 days after the first dose, and were maintained between weekly 1 mg/kg SC doses. Following discontinuation of RAPTIVA, CD11a expression returned to a mean of 74% of baseline at 5 weeks and stayed at comparable levels at 8 and 13 weeks. Following discontinuation of RAPTIVA, free CD11a binding sites returned to a mean of 86% of baseline at 8 weeks and stayed at comparable levels at 13 weeks. No assessments of CD11a expression or free CD11a binding sites were made after 13 weeks.
In clinical trials, RAPTIVA treatment resulted in a mean increase (relative to baseline) in white blood cell (WBC) count of 34%, a doubling of mean lymphocyte counts and an increase in eosinophil counts of 29% due to decreased leukocyte adhesion to blood vessel walls and decreased trafficking from the vascular compartment to tissues. At Day 56 of 1 mg/kg/wk RAPTIVA treatment, 32% (213/676) of patients had a shift in total WBC from low or normal baseline value to above normal, 46% (324/701) had a shift to above normal absolute lymphocyte counts, and 5% (35/675) had a shift to above normal eosinophil counts. Following discontinuation of RAPTIVA treatment, the abnormal elevated lymphocyte counts took approximately 8 weeks to normalize among patients who had above normal lymphocyte counts. Plasma samples collected after first administration of 0.3 mg/kg IV RAPTIVA indicate that at 2 hours TNF-(alpha) and IL-6 plasma levels were elevated 9- and 90-fold, respectively, compared with baseline. Plasma samples collected after first administration of 0.7 mg/kg SC RAPTIVA indicate that at 2 days, IL-6 levels were elevated (10 pg/mL as compared with 5 pg/mL at baseline), whereas TNF-(alpha) was not detectable. In RAPTIVA-treated patients the mean levels of C reactive protein increased from baseline by 67% and the mean levels of fibrinogen increased by 15%.
CLINICAL STUDIES
RAPTIVA was evaluated in four randomized, double-blind, placebo-controlled studies in adults with chronic (>6 months), stable, plaque psoriasis, who had a minimum body surface area involvement of 10% and who were candidates for, or had previously received systemic therapy or phototherapy. In these studies 54-70% of patients had previously received systemic therapy or phototherapy (PUVA) for psoriasis. Patients with clinically significant flares and patients with guttate, erythrodermic, or pustular psoriasis as the sole form of psoriasis were excluded from the studies. Patients were randomized to receive doses of 1 mg/kg or 2 mg/kg of RAPTIVA or placebo administered once a week for 12 weeks. Patients randomized to RAPTIVA received 0.7 mg/kg as the first dose prior to receiving the full assigned dose in subsequent weeks. During the studies, patients could receive concomitant low potency topical steroids. No other concomitant psoriasis therapies were allowed during treatment or the follow-up period.
Patients were evaluated using the Psoriasis Area and Severity Index (PASI) during the study. The PASI is a composite score that takes into consideration both the fraction of body surface area affected and the nature and severity of the psoriatic changes within the affected regions (erythema, infiltration/plaque thickness, and desquamation). Both treatment groups in all four studies had baseline median PASI scores of 17. Both treatment groups across all four studies had baseline median body surface area involvement ranging between 22-28%. Compared with placebo, more patients randomized to RAPTIVA had at least a 75% reduction from baseline PASI score (PASI-75) 1 week after the 12-week treatment period (Table 1). RAPTIVA 2 mg/kg was not superior to RAPTIVA 1 mg/kg.
Table 1
Proportion of Patients with >/=75% Improvement
in PASI after 12 Weeks of Treatment (PASI-75)Placebo RAPTIVA
1 mg/kg/wkDifference
(95%CI)Study 14%
n = 18727% a
n = 36922%
(16%, 29%)Study 22%
n = 17039% a
n = 16237%
(28%, 46%)Study 35%
n = 12222% a
n = 23217%
(9%, 27%)Study 43%
n = 23624% a
n = 45021%
(15%, 27%)a p<0.001 for comparison of RAPTIVA group with placebo group using Fisher's exact test within each study.
All three components of the PASI (plaque induration, scaling, and erythema) contributed comparably to the improvement in PASI. Other clinical responses evaluated (Table 2) included the proportion of patients who achieved minimal or clear status by a static Physician Global Assessment (sPGA) and the proportion of patients with a reduction in PASI of at least 50% from baseline (PASI-50) 1 week following the 12-week treatment period. The sPGA is a 6 category scale ranging from "very severe" to "clear" indicating the physician's overall assessment of the psoriasis severity focusing on plaque, scaling and erythema. Treatment success of minimal or clear consisted of none or slight elevation in plaque, none or minimal white color in scaling, and up to moderate definite red coloration in erythema. Across all four studies, the percentage of patients with baseline sPGA classifications of moderate was 48-56%, severe 33-43%, and 3-6% were classified as very severe.
Table 2
Percentage of Patients # Responding after 12 Weeks of TreatmentOutcome MeasurementStudyPlaceboRAPTIVA
1 mg/kg/wkDifference a
(95% CI)sPGA: Minimal or Clear1 3% 26% 23% (16, 30) 2 3% 32% 29% (21, 39) 3 3% 19% 16% (8, 25) 4 4% 20% 16% (11, 22) >50% improvement in PASI (PASI-50)1 14% 59% 45% (37, 53) 215% 61% 46% (37, 56) 3 16% 52% 36% (26, 47) 4 14% 52% 38% (31, 45) The number of patients in each study and treatment group is the same as listed in Table 1.a p <0.001 for comparison of RAPTIVA group to placebo group using Fisher's exact test for all comparisons between groups.
In Study 1, 12% of RAPTIVA-treated patients achieved a PASI-50 at Week 4 compared with 5% for placebo. The median time to PASI-50 among PASI-75 achievers was approximately 6 weeks. Similar results were observed in Studies 2, 3, and 4.
In Study 3, sustained response to extended RAPTIVA treatment was evaluated. RAPTIVA-treated patients who achieved a PASI-75 response at Week 12 were re-randomized to receive RAPTIVA or placebo for a second contiguous 12-week treatment period. Sixty-one of 79 patients (77%) re-randomized to a second 12-week treatment period with RAPTIVA maintained PASI-75 response compared with 8 of 40 patients (20%) re-randomized to placebo. Sustained responses to RAPTIVA have also been observed in uncontrolled, open-label extension treatment trials when patients received RAPTIVA without interruption for 24 weeks.
In Study 2, response to intermittent RAPTIVA treatment was evaluated among patients who achieved PASI-75 response with 12 weeks of RAPTIVA treatment and were followed off-treatment until relapse of psoriasis (50% loss of treatment response). In patients who resumed RAPTIVA treatment upon relapse of psoriasis, 31% (17/55) re-established a PASI-75 response (compared with the initial baseline). After 12 weeks of treatment, the median duration of a PASI-75 response after RAPTIVA discontinuation was between 1 and 2 months.
The safety and efficacy of RAPTIVA therapy beyond 1 year have not been established.
INDICATIONS AND USAGE
RAPTIVA® [efalizumab] is indicated for the treatment of adult patients (18 years or older) with chronic moderate to severe plaque psoriasis who are candidates for systemic therapy or phototherapy.
CONTRAINDICATIONS
RAPTIVA should not be administered to patients with known hypersensitivity to RAPTIVA or any of its components.
WARNINGS
Serious Infections
RAPTIVA is an immunosuppressive agent and has the potential to increase the risk of infection and reactivate latent, chronic infections. RAPTIVA should not be administered to patients with clinically important infections. Caution should be exercised when considering the use of RAPTIVA in patients with a chronic infection or history of recurrent infections. If a patient develops a serious infection, RAPTIVA should be discontinued. New infections developing during RAPTIVA treatment should be monitored. During the first 12 weeks of controlled trials, serious infections occurred in 7 of 1620 (0.4 %) RAPTIVA-treated patients compared with 1 of 715 (0.1%) placebo-treated patients (see ADVERSE REACTIONS , Infections ). Serious infections requiring hospitalization included cellulitis, pneumonia, abscess, sepsis, bronchitis, gastroenteritis, aseptic meningitis, Legionnaire's disease, and vertebral osteomyelitis (note some patients had more than one infection). Postmarketing reports of serious infections include necrotizing fasciitis and tuberculous pneumonia. Bacterial sepsis with seeding of distant sites, severe pneumonia with neutropenia (ANC 60/mm 3 ), and worsening of infection (e.g., cellulitis, pneumonia) despite antimicrobial treatment have been observed.
Malignancies
RAPTIVA is an immunosuppressive agent. Many immunosuppressive agents have the potential to increase the risk of malignancy. The role of RAPTIVA in the development of malignancies is not known. Caution should be exercised when considering the use of RAPTIVA in patients at high risk for malignancy or with a history of malignancy. If a patient develops a malignancy, RAPTIVA should be discontinued (see ADVERSE REACTIONS , Malignancies ).
Immune-Mediated Thrombocytopenia
Platelet counts at or below 52,000 cells per µL were observed in 8 (0.3%) RAPTIVA-treated patients during clinical trials compared with none among the placebo-treated patients (see ADVERSE REACTIONS , Immune-Mediated Thrombocytopenia ). Five of the 8 patients received a course of systemic steroids for thrombocytopenia. Thrombocytopenia resolved in the 7 patients receiving adequate follow-up (1 patient was lost to follow-up). Reports of severe thrombocytopenia have also been received postmarketing. Physicians should follow patients closely for signs and symptoms of thrombocytopenia. Assessment of platelet counts is recommended during treatment with RAPTIVA (see PRECAUTIONS , Laboratory Tests ) and RAPTIVA should be discontinued if thrombocytopenia develops.
Immune-Mediated Hemolytic Anemia
Reports of hemolytic anemia, some serious, diagnosed 4-6 months after the start RAPTIVA treatment have been received. RAPTIVA should be discontinued if hemolytic anemia occurs.
Psoriasis Worsening and Variants
Worsening of psoriasis can occur during or after discontinuation of RAPTIVA. During clinical studies, 19 of 2589 (0.7%) of RAPTIVA-treated patients had serious worsening of psoriasis during treatment (n = 5) or worsening past baseline after discontinuation of RAPTIVA (n = 14) (see ADVERSE REACTIONS , Adverse Events of Psoriasis ). In some patients these events took the form of psoriatic erythroderma, pustular psoriasis, or development of new plaque lesions. Some patients required hospitalization and alternative antipsoriatic therapy to manage the psoriasis worsening. Patients, including those not responding to RAPTIVA treatment, should be closely observed following discontinuation of RAPTIVA, and appropriate psoriasis treatment instituted as necessary.
PRECAUTIONS
Arthritis Events
Infrequent new onset or recurrent severe arthritis events, including psoriatic arthritis events, have been reported in clinical trials and postmarketing. These arthritis events began while on treatment or following discontinuation of RAPTIVA and were uncommonly associated with flare of psoriasis. Patients improved after discontinuation of RAPTIVA with or without anti-arthritis therapy.
Immunosuppression
The safety and efficacy of RAPTIVA in combination with other immunosuppressive agents or phototherapy have not been evaluated. Patients receiving other immunosuppressive agents should not receive concurrent therapy with RAPTIVA because of the possibility of increased risk of infections and malignancies.
Immunizations
The safety and efficacy of vaccines, administered to patients being treated with RAPTIVA have not been studied. In a small clinical study with IV administered RAPTIVA, a single dose of 0.3 mg/kg given before primary immunization with a neoantigen decreased the secondary immune response, and a dose of 1 mg/kg almost completely ablated it. A dose of 0.3 mg/kg IV has comparable pharmacodynamic effects to the recommended dose of 1 mg/kg SC. In chimpanzees exposed to RAPTIVA at >/=10 times the clinical exposure level (based on mean peak plasma levels) antibody responses were decreased following immunization with tetanus toxoid compared with untreated control animals. Acellular, live and live-attenuated vaccines should not be administered during RAPTIVA treatment.
First Dose Reactions
First dose reactions including headache, fever, nausea, and vomiting are associated with RAPTIVA treatment and are dose-level related in incidence and severity (see ADVERSE REACTIONS ). Therefore, a conditioning dose of 0.7 mg/kg is recommended to reduce the incidence and severity of reactions associated with initial dosing (see DOSAGE AND ADMINISTRATION ). Cases of aseptic meningitis resulting in hospitalization have been observed in association with initial dosing (see ADVERSE REACTIONS , Inflammatory/Immune-Mediated Reactions ).
Information for Patients
Patients should be informed that their physician may monitor platelet counts during therapy. Patients should be advised to seek immediate medical attention if they develop any of the signs and symptoms associated with severe thrombocytopenia (such as easy bleeding from the gums, bruising, or petechiae) or with severe hemolytic anemia (such as weakness, orthostatic light-headedness, hemoglobinuria or jaundice), or with worsening of psoriasis or arthritis. Patients should also be informed that RAPTIVA is an immunosuppressant, and could increase their chances of developing an infection or a malignancy. Patients should be advised to promptly call the prescribing doctor's office if they develop any new signs of, or receive a new diagnosis of infection or malignancy while undergoing treatment with RAPTIVA.
Female patients should also be advised to notify their physicians if they become pregnant while taking RAPTIVA (or within 6 weeks of discontinuing RAPTIVA) and be advised of the existence of and encouraged to enroll in the RAPTIVA Pregnancy Registry. Call 1-877-RAPTIVA (1-877-727-8482) to enroll into the Registry.
If a patient or caregiver is to administer RAPTIVA, he/she should be instructed regarding injection techniques and how to measure the correct dose to ensure proper administration of RAPTIVA. Patients should be also referred to the RAPTIVA Patient Package Insert. In addition, patients should have available materials for and be instructed in the proper disposal of needles and syringes to comply with state and local laws. Patients should also be cautioned against reuse of syringes and needles.
Laboratory Tests
Assessment of platelet counts is recommended upon initiating and periodically while receiving RAPTIVA treatment. It is recommended that assessments be more frequent when initiating therapy (e.g., monthly) and may decrease in frequency with continued treatment (e.g., every 3 months). Severe thrombocytopenia has been observed (see WARNINGS , Immune-Mediated Thrombocytopenia ).
Drug Interactions
No formal drug interaction studies have been per-formed with RAPTIVA. RAPTIVA should not be used with other immunosuppressive drugs (see PRECAUTIONS , Immunosuppression ).
Acellular, live and live-attenuated vaccines should not be administered during RAPTIVA treatment (see PRECAUTIONS , Immunizations ).
Drug/Laboratory Test Interactions
Increases in lymphocyte counts related to the pharmacologic mechanism of action are frequently observed during RAPTIVA treatment (see CLINICAL PHARMACOLOGY , Pharmacodynamics ).
Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term animal studies have not been conducted to evaluate the carcinogenic potential of RAPTIVA.
Subcutaneous injections of male and female mice with an anti-mouse CD11a antibody at up to 30 times the equivalent of the 1 mg/kg clinical dose of RAPTIVA had no adverse effects on mating, fertility, or reproduction parameters. The clinical significance of this observation is uncertain.
Genotoxicity studies were not conducted.
Pregnancy (Category C)
Animal reproduction studies have not been conducted with RAPTIVA. It is also not known whether RAPTIVA can cause fetal harm when administered to a pregnant woman or can affect reproduction capacity. RAPTIVA should be given to a pregnant woman only if clearly needed.
In a developmental toxicity study conducted in mice using an anti-mouse CD11a antibody at up to 30 times the equivalent of the recommended clinical dose of RAPTIVA, no evidence of maternal toxicity, embryotoxicity, or teratogenicity was observed when administered during organogenesis. No adverse effects on behavioral, reproductive, or growth parameters were observed in offspring of female mice subcutaneously treated with an anti-mouse CD11a antibody during gestation and lactation using doses 3- to 30-times the equivalent of the recommended clinical dose of RAPTIVA. At 11 weeks of age, the offspring of these females exhibited a significant reduction in their ability to mount an antibody response, which showed evidence of partial reversibility by 25 weeks of age. Animal studies, however, are not always predictive of human response, and there are no adequate and well-controlled studies in pregnant women.
Since the effects of RAPTIVA on pregnant women and fetal development, including immune system development are not known, healthcare providers are encouraged to enroll patients who become pregnant while taking RAPTIVA (or within 6 weeks of discontinuing RAPTIVA) in the RAPTIVA Pregnancy Registry by calling 1-877-RAPTIVA (1-877-727-8482).
Nursing Mothers
It is not known whether RAPTIVA is excreted in human milk. An anti-mouse CD11a antibody was detected in milk samples of lactating mice exposed to anti-mouse CD11a antibody and the offspring of the exposed females exhibited significant reduction in antibody responses (see PRECAUTIONS , Pregnancy ). Since maternal immunoglobulins are known to be present in the milk of lactating mothers, and animal data suggest the potential for adverse effects in nursing infants from RAPTIVA, a decision should be made whether to discontinue nursing while taking the drug or to discontinue the use of the drug, taking into account the importance of the drug to the mother.
Pediatric Use
The safety and efficacy of RAPTIVA in pediatric patients have not been studied.
Geriatric Use
Of the 1620 patients who received RAPTIVA in controlled trials, 128 were >/=65 years of age, and 2 were >/=75 years of age. Although no differences in safety or efficacy were observed between older and younger patients, the number of patients aged 65 and over is not sufficient to determine whether they respond differently from younger patients. Because the incidence of infections is higher in the elderly population, in general, caution should be used in treating the elderly.
ADVERSE REACTIONS
The most serious adverse reactions observed during treatment with RAPTIVA were serious infections, malignancies, thrombocytopenia, hemolytic anemia, arthritis events, and psoriasis worsening and variants (see WARNINGS ).
The most common adverse reactions associated with RAPTIVA were a first dose reaction complex that included headache, chills, fever, nausea, and myalgia within two days following the first two injections. These reactions are dose-level related in incidence and severity and were largely mild to moderate in severity when a conditioning dose of 0.7 mg/kg was used as the first dose. In placebo-controlled trials, 29% of patients treated with RAPTIVA 1 mg/kg developed one or more of these symptoms following the first dose compared with 15% of patients receiving placebo. After the third dose, 4% and 3% of patients receiving RAPTIVA 1 mg/kg and placebo, respectively, experienced these symptoms. Less than 1% of patients discontinued RAPTIVA treatment because of these adverse events.
Other adverse events resulting in discontinuation of RAPTIVA treatment were psoriasis (0.6%), pain (0.4%), arthritis (0.4%), and arthralgia (0.3%).
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of one drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data described below reflect RAPTIVA exposure for 2762 adult psoriasis patients (age range 18 to 75 years), including 2400 patients exposed for three months, 904 for six months, and 218 exposed for one year or more, in all controlled and uncontrolled studies. The median age of patients receiving RAPTIVA was 44 years, with 189 patients above the age of 65; 67% were men, and 89% were Caucasian. These data include patients treated at doses higher than the recommended dose of 1 mg/kg weekly.
Controlled clinical trials provide the most informative basis for estimating the frequency of RAPTIVA-related adverse drug reactions. Table 3 enumerates the adverse events occurring during controlled periods of the clinical trials where the frequency of the adverse events is at least 2% greater in the RAPTIVA-treated group than the placebo group.
Table 3
Adverse Events in Placebo Controlled Study Periods
Reported at a >/=2% Higher Rate in the 1 mg/kg/wk
RAPTIVA Treatment than Placebo GroupsPlacebo
(n = 715)RAPTIVA
1 mg/kg/wk
(n = 1213)Headache159 (22%) 391 (32%) Infection a188 (26%) 350 (29%) Chills32 (4%) 154 (13%) Nausea51 (7%) 128 (11%) Pain38 (5%) 122 (10%) Myalgia35 (5%) 102 (8%) Flu Syndrome29 (4%) 83 (7%) Fever24 (3%) 80 (7%) Back pain14 (2%) 50 (4%) Acne4 (1%) 45 (4%) a Includes diagnosed infections and other non-specific infections. Most common non-specific infection was upper respiratory infection.
Adverse events occurring at a rate between 1 and 2% greater in the RAPTIVA group compared with placebo were arthralgia, asthenia, peripheral edema, and psoriasis.
The following serious adverse reactions were observed in RAPTIVA-treated patients.
Infections
In the first 12 weeks of placebo-controlled studies, the proportion of patients with serious infection was 0.4% (7/1620) in the RAPTIVA-treated group (5 of these were hospitalized, 0.3%) and 0.1% (1/715) in the placebo group (see WARNINGS , Serious Infections ). In the complete safety data from both controlled and uncontrolled studies, the overall incidence of hospitalization for infections was 1.6 per 100patient-years for RAPTIVA-treated patients compared with 1.2 per 100 patient-years for placebo-treated patients. Including both controlled, uncontrolled, and follow-up study treatment periods there were 27 serious infections in 2475 RAPTIVA-treated patients. These infections included cellulitis, pneumonia, abscess, sepsis, sinusitis, bronchitis, gastroenteritis, aseptic meningitis, Legionnaire's disease, septic arthritis, and vertebral osteomyelitis. In controlled trials, the overall rate of infections in RAPTIVA-treated patients was 3% higher than in placebo-treated patients (Table 3).
Malignancies
Among the 2762 psoriasis patients who received RAPTIVA at any dose (median duration 8 months), 31 patients were diagnosed with 37 malignancies (see WARNINGS , Malignancies ). The overall incidence of malignancies of any kind was 1.8 per 100 patient-years for RAPTIVA-treated patients compared with 1.6 per 100 patient-years for placebo-treated patients. Malignancies observed in the RAPTIVA-treated patients included non-melanoma skin cancer, non-cutaneous solid tumors, Hodgkin's lymphoma and non-Hodgkin's lymphoma, and malignant. The incidence of non-cutaneous solid tumors (8 in 1790 patient-years) and malignant melanoma were within the range expected for the general population.
The majority of the malignancies were non-melanoma skin cancers; 26 cases (13 basal, 13 squamous) in 20 patients (0.7% of 2762 RAPTIVA-treated patients). The incidence was comparable for RAPTIVA-treated and placebo-treated patients. However, the size of the placebo group and duration of follow-up were limited and a difference in rates of non-melanoma skin cancers cannot be excluded.
Immune-Mediated Thrombocytopenia
In the combined safety database of 2762 RAPTIVA-treated patients, there were eight occurrences (0.3%) of thrombocytopenia of <52,000 cells per µL reported (see WARNINGS , Immune-Mediated Thrombocytopenia ). Three of the eight patients were hospitalized for thrombocytopenia, including one patient with heavy uterine bleeding; all cases were consistent with an immune mediated thrombocytopenia. Antiplatelet antibody was evaluated in one patient and was found to be positive. Each case resulted in discontinuation of RAPTIVA. Based on available platelet count measurements, the onset of platelet decline was between 8 and 12 weeks after the first dose of RAPTIVA in 5 of the patients. Onset was more delayed in 3 patients, occurring as late as one year in 1 patient. In these cases, the platelet count nadirs occurred between 12 and 72 weeks after the first dose of RAPTIVA.
Immune-Mediated Hemolytic Anemia
Two reports of hemolytic anemia were observed in clinical trials. Additional cases were reported in the postmarketing setting. The anemia was diagnosed 4-6 months after the start of RAPTIVA and in two serious cases the hemoglobin level decreased to 6 and 7 g/dl. RAPTIVA treatment was discontinued, erythrocyte transfusions and other therapies were administered (see WARNINGS , Immune-Mediated Hemolytic Anemia ).
Adverse Events of Psoriasis
In the combined safety database from all studies, serious psoriasis adverse events occurred in 19 RAPTIVA-treated patients (0.7%) including hospitalization in 17 patients (see WARNINGS , Psoriasis Worsening and Variants ). Most of these events (14/19) occurred after discontinuation of study drug and occurred in both patients responding and not responding to RAPTIVA treatment. Serious adverse events of psoriasis included pustular, erythrodermic, and guttate subtypes. During the first 12 weeks of treatment within placebo-controlled studies, the rate of psoriasis adverse events (serious and non-serious) was 3.2% (52/1620) in the RAPTIVA-treated patients and 1.4% (10/715) in the placebo-treated patients.
Arthritis Events
Infrequent new onset or recurrent severe arthritis events, including psoriatic arthritis events, have been reported in clinical trials and postmarketing (see PRECAUTIONS , Arthritis Events ).
Hypersensitivity Reactions
Symptoms associated with a hypersensitivity reaction (e.g., dyspnea, asthma, urticaria, angioedema, maculopapular rash) were evaluated by treatment group. In the first 12 weeks of the controlled clinical studies, the proportion of patients reporting at least one hypersensitivity reaction was 8% (95/1213) in the 1 mg/kg/wk group and 7% (49/715) patients in the placebo group. Urticaria was observed in 1% of patients (16/1213) receiving RAPTIVA and 0.4% of patients (3/715) receiving placebo during the initial 12-week treatment period. Other observed adverse events in patients receiving RAPTIVA that may be indicative of hypersensitivity included: laryngospasm, angioedema, erythema multiforme, asthma, and allergic drug eruption. One patient was hospitalized with a serum sickness-like reaction.
Inflammatory/Immune-Mediated Reactions
In the entire RAPTIVA clinical development program of 2762 RAPTIVA-treated patients, inflammatory, potentially immune-mediated adverse events resulting in hospitalization included inflammatory arthritis (12 cases, 0.4% of patients) and interstitial pneumonitis (2 cases). One case each of the following serious adverse reactions was observed: transverse myelitis, bronchiolitis obliterans, aseptic meningitis, idiopathic hepatitis, sialedenitis, and sensorineural hearing loss. Myositis, eosinophilic pneumonitis, resolving after discontinuation of RAPTIVA have been reported postmaketing.
Postmarketing Experience
In postmarketing experience, other reported adverse events included toxic epidermal necrolysis and photosensitivity reactions.
Laboratory Values
In RAPTIVA-treated patients, a mean elevation in alkaline phosphatase (5 Units/L) was observed; 4% of RAPTIVA-treated patients experienced a shift to above normal values compared with 0.6% of placebo-treated patients. The clinical significance of this change is unknown. Higher numbers of RAPTIVA-treated patients experienced elevations above normal in two or more liver function tests than placebo (3.1% vs. 1.5%).
Other laboratory adverse reactions that were observed included thrombocytopenia, (see WARNINGS , and ADVERSE REACTIONS , Immune-Mediated Thrombocytopenia ), lymphocytosis (40%) (including three cases of transient atypical lymphocytosis), and leukocytosis (26%).
Immunogenicity
In patients evaluated for antibodies to RAPTIVA after RAPTIVA treatment ended, predominantly low-titer antibodies to RAPTIVA or other protein components of the RAPTIVA drug product were detected in 6.3% (67/1063) of patients. The long-term immunogenicity of RAPTIVA is unknown.
The data reflect the percentage of patients whose test results were considered positive for antibodies to RAPTIVA in the ELISA assay, and are highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody positivity in an assay may be influenced by several factors including sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to RAPTIVA with the incidence of antibodies to other products may be misleading.
OVERDOSAGE
Doses up to 4 mg/kg/wk SC for 10 weeks following a conditioning (0.7 mg/kg) first dose have been administered without an observed increase in acute toxicity. The maximum administered single dose was 10 mg/kg IV. This was administered to one patient, who subsequently was admitted to the hospital for severe vomiting. In case of overdose, it is recommended that the patient be monitored for 24-48 hours for any acute signs or symptoms of adverse reactions or effects and appropriate treatment instituted.
DOSAGE AND ADMINISTRATION
The recommended dose of RAPTIVA® [efalizumab] is a single 0.7 mg/kg SC conditioning dose followed by weekly SC doses of 1 mg/kg (maximum single dose not to exceed a total of 200 mg).
RAPTIVA is intended for use under the guidance and supervision of a physician. If it is determined to be appropriate, patients may self-inject RAPTIVA after proper training in the preparation and injection technique and with medical follow-up.
Preparation for Administration
RAPTIVA should be administered using the sterile, disposable syringe and needles provided (see HOW SUPPLIED section). Remove the cap from the pre-filled syringe containing sterile water for injection (non-USP) and attach the needle to the syringe. Remove the plastic cap protecting the rubber stopper of the RAPTIVA vial and wipe the top of the rubber stopper with one of the provided alcohol swabs. After cleaning with the alcohol swab, do not touch the top of the vial. To prepare the RAPTIVA solution, using the provided pre-filled diluent syringe slowly inject the 1.3 mL of sterile water for injection (non-USP) into the RAPTIVA vial. Swirl the vial with a GENTLE rotary motion to dissolve the product. DO NOT SHAKE. Shaking will cause foaming of the RAPTIVA solution. Generally, dissolution of RAPTIVA takes less than 5 minutes. RAPTIVA is provided as a single-use vial and contains no antibacterial preservatives. Reconstitute immediately before use and use only once. If the reconstituted RAPTIVA is not used immediately, store the RAPTIVA vial at room temperature and use within 8 hours. The reconstituted solution should be clear to pale yellow and free of particulates.
Administration
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to subcutaneous administration. If particulates or discolorations are noted, the product should not be used. Insert the needle into the vial containing the RAPTIVA solution, invert the vial, and keeping the needle below the level of the liquid, withdraw the dose to be given into the syringe. Replace the needle on the syringe with a new needle.
No other medications should be added to solutions containing RAPTIVA, and RAPTIVA should not be reconstituted with other diluents.
Sites for injection include thigh, abdomen, buttocks, or upper arm. Injection sites should be rotated.
Following administration, discard any unused reconstituted RAPTIVA solution.
Stability and Storage
Do not use a vial beyond the expiration date stamped on the carton or vial label. RAPTIVA (lyophilized powder) must be refrigerated at 2-8°C (36-46°F). Protect the vial from exposure to light. Store in original carton until time of use.
HOW SUPPLIED
RAPTIVA® [efalizumab] is supplied as a lyophilized, sterile powder to deliver 125 mg of efalizumab per single-use vial.
Each RAPTIVA carton contains four trays. Each tray contains one single-use vial designed to deliver 125 mg of efalizumab, one single-use prefilled diluent syringe containing 1.3 mL sterile water for injection (non-USP), two 25 gauge × 5/8 inch needles, two alcohol prep pads, a package insert with an accompanying patient information insert. The NDC number for the four administration dose pack carton is 50242-058-04.
REFERENCES
- Werther WA, Gonzalez TN, O'Connor SJ, McCabe S, Chan B, Hotaling T, et al. Humanization of an anti-lymphocyte function-associated antigen (LFA)-1 monoclonal antibody and reengineering of the humanized antibody for binding to rhesus LFA-1. J Immunol 1996;157:4986-95.
Patient Information
RAPTIVA (Rap-TEE-vah)
[efalizumab]
for injection, subcutaneous
Read the Patient Information that comes with RAPTIVA® [efalizumab] before you start using it and each time you get a refill. There may be new information. This information does not take the place of talking with your healthcare provider about your medical condition or treatment. It is important to remain under a healthcare provider's care while using RAPTIVA. Do not change or stop treatment without first talking with your healthcare provider. Talk to your healthcare provider or pharmacist if you have any questions about RAPTIVA.
WHAT IS THE MOST IMPORTANT INFORMATION I SHOULD KNOW ABOUT RAPTIVA?
RAPTIVA can decrease the activity of your immune system. Therefore, people using RAPTIVA may have an increased chance of getting:
- Serious infections. Some infections could become serious. If you have an infection, tell your healthcare provider before you start using RAPTIVA. If you get an infection that does not go away while taking RAPTIVA, tell your healthcare provider right away.
- Cancers. Many drugs that decrease the activity of the immune system can increase the risk of cancer. If you have had cancer you should tell your healthcare provider before you start taking RAPTIVA. The role of RAPTIVA in the development of cancer is not known.
- Low platelet counts (thrombocytopenia). Platelets help your blood clot. Low platelets give you a higher chance for bleeding. Call your doctor right away if you have increased bruising or bleeding. Your healthcare provider may do regular blood tests to check your platelets while you are taking RAPTIVA.
- Low blood counts (anemia). RAPTIVA may increase the breakdown of your red blood cells and cause very low blood counts. Call your doctor right away if you feel weak and lightheaded, your skin and eyes turn yellow in color or your urine turns red or dark.
- Worsening of psoriasis. Some patients have had severe worsening or new forms of psoriasis while taking RAPTIVA or after stopping RAPTIVA. Tell your healthcare provider right away if your psoriasis gets worse or if you see any new rashes during or after treatment with RAPTIVA.
- Arthritis. Some patients have had worsening or new arthritis while taking RAPTIVA or after stopping RAPTIVA. Tell your health care provider if you have severe redness, pain, swelling, or stiffness of joints such as hands, knees, ankles, etc.
You should not receive vaccines while using RAPTIVA. RAPTIVA may prevent a vaccine from working. Talk to your healthcare provider if you need to receive a vaccine while using RAPTIVA.
WHAT IS RAPTIVA?
RAPTIVA is a medicine used to treat adult patients with moderate to severe plaque psoriasis who can be treated with medicines that affect the whole body (systemic therapy) or with phototherapy.
RAPTIVA is a man-made protein that is like proteins made in the body called antibodies. Antibodies fight disease in the human body. RAPTIVA may decrease the skin changes in the body that are the main problems of moderate to severe plaque psoriasis.
RAPTIVA has not been studied in children under 18 years of age.
WHO SHOULD NOT USE RAPTIVA?
Do not use RAPTIVA if you have ever had an allergic reaction to RAPTIVA.
Before using RAPTIVA, tell your healthcare provider
-
about the following medical conditions:
- If you are pregnant, planning to become pregnant, or become pregnant while using RAPTIVA. It is not known if RAPTIVA can harm your unborn baby. If you become pregnant while taking RAPTIVA, notify your healthcare provider immediately. You and your healthcare provider will have to decide if RAPTIVA is right for you during pregnancy. If you use RAPTIVA when you are pregnant, call 1-877-RAPTIVA (1-877-727-8482) to ask how you can be included in the RAPTIVA Pregnancy Registry.
- If you are breast feeding. It is not known if RAPTIVA passes into your milk. It may harm your baby. You will need to decide whether to use RAPTIVA or breast feed, but you may not do both.
- If you have any infections (see WHAT IS THE MOST IMPORTANT INFORMATION I SHOULD KNOW ABOUT RAPTIVA? ).
- If you have immune system problems
-
about all the medicines you take including prescription and nonprescription medicines, vitamins, and herbal supplements.
It is not known if RAPTIVA and other medicines affect each other.
Especially, tell your healthcare provider if you are using:
- Other medicines or treatments for your psoriasis
- Medicines called immunosuppressives or any medicine that affects your immune system. Ask your healthcare provider or pharmacist if you are not sure if any of your medicines are immunosuppressives.
HOW SHOULD I USE RAPTIVA?
- RAPTIVA is an injection that you give yourself once a week.
- See the end of this leaflet for instructions on how to prepare and inject RAPTIVA ( HOW DO I PREPARE AND GIVE A RAPTIVA INJECTION? ). Ask your healthcare provider or pharmacist if you have any questions about using RAPTIVA.
- Use RAPTIVA exactly as prescribed by your healthcare provider. Your dose of RAPTIVA is based on your body weight. Tell your healthcare provider if your weight changes. Do not change your dose without talking to your healthcare provider. Do not stop using RAPTIVA without talking to your healthcare provider.
- RAPTIVA is injected under the skin (subcutaneous) of your upper leg (thigh), upper arm, abdomen, or buttocks once a week. Change (rotate) your skin injection site with each injection.
- Use RAPTIVA the same day each week. If you miss your dose of RAPTIVA contact your healthcare provider to find out when to take your next dose of RAPTIVA and what schedule to follow after that.
- If you take more than your regular dose of RAPTIVA, call your healthcare provider right away.
- See your healthcare provider regularly while using RAPTIVA. Do not miss your appointments. Your healthcare provider may do blood tests including platelet counts before and during treatment with RAPTIVA to check its affect on your body.
WHAT SHOULD I AVOID WHILE USING RAPTIVA?
Unless directed by your healthcare provider, do not:
- take other medicines called immunosuppressives.
- take treatments called phototherapy.
You should not receive vaccines while using RAPTIVA. Talk to your healthcare provider if you need to receive a vaccine while taking RAPTIVA (see WHAT IS THE MOST IMPORTANT INFORMATION I SHOULD KNOW ABOUT RAPTIVA? ).
WHAT ARE THE POSSIBLE SIDE EFFECTS OF RAPTIVA?
RAPTIVA can cause serious side effects including the following (see WHAT IS THE MOST IMPORTANT INFORMATION I SHOULD KNOW ABOUT RAPTIVA?):
RAPTIVA can affect your immune system and might cause:
- Serious infections
- Cancers
- Low platelet counts (thrombocytopenia)
- Low blood counts (anemia)
- Worsening of psoriasis
- New or worsening arthritis
The most common side effects of RAPTIVA include headache, chills, fever, nausea, and muscle aches. These reactions usually happen within the first 48 hours following RAPTIVA injection, and often decrease after the first few weeks of use of RAPTIVA. Other side effects that can also happen with RAPTIVA include back pain, or swelling of the arms or legs (peripheral edema). Talk to your healthcare provider about any symptoms that bother you.
If you get any side effect that concerns you or if you get an infection, call your healthcare provider.
These are not all the side effects of RAPTIVA. For more information, ask your healthcare provider or pharmacist.
HOW SHOULD I STORE RAPTIVA?
-
Store RAPTIVA vials in the refrigerator at 36° to 46°F (2° to 8°C) until you are ready to prepare your injection.
Do not freeze or store at room temperature.
Once RAPTIVA has been mixed with sterile water, you should use it right away to inject yourself. If you are unable to inject the drug after mixing, the mixture can stay at room temperature for up to 8 hours. Do not use RAPTIVA that was mixed more than 8 hours earlier.
If you are traveling, be sure to store RAPTIVA at the right temperature. If you have any questions, ask your healthcare provider or pharmacist. - Protect RAPTIVA vials from light while stored.
- Throw away RAPTIVA vials that are out of date.
- Keep RAPTIVA and all medicines out of the reach of children.
GENERAL INFORMATION ABOUT RAPTIVA
Medicines are sometimes prescribed for conditions that are not mentioned in patient information leaflets. Do not use RAPTIVA for a condition for which it was not prescribed. Do not give RAPTIVA to other people, even if they have the same symptoms you have. It may harm them.
This leaflet summarizes the most important information about RAPTIVA. If you would like more information, talk with your healthcare provider. You can ask your healthcare provider or pharmacist for information about RAPTIVA that is written for health professionals. For more information, you can also call 1-877-RAPTIVA (toll free).
HOW DO I PREPARE AND GIVE A RAPTIVA INJECTION?
If your dose amount is more than 1.25 mL, you will need to use 2 RAPTIVA blister trays, and you will give yourself 2 injections of RAPTIVA.
Setting Up the Equipment
- Take the RAPTIVA® (efalizumab) blister tray out of the refrigerator, and place it on a flat, well-lit, clean work surface.
- Wash your hands with soap and water before opening the blister tray.
-
Open the tray and lay out the contents. Allow the contents to come to room temperature.
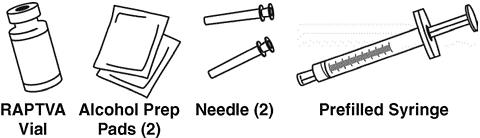
As shown below, the tray contains:
- One RAPTIVA vial
- One 1.3-mL prefilled syringe of sterile water
- Two 25-gauge needles
- Two alcohol prep pads
Contact your healthcare provider or pharmacist if you are missing any of the items listed above.
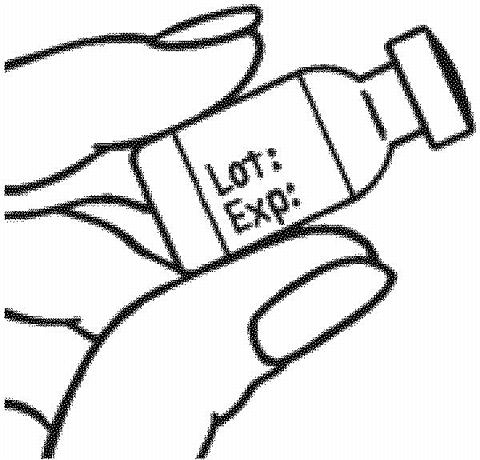
- Check the expiration (Exp.) date on the RAPTIVA vial label and prefilled syringe label. If the expiration date has passed, do not use the RAPTIVA vial or the prefilled syringe containing the sterile water. Contact your healthcare provider.
-
Partially peel open the needle pack and place it on a clean surface. Be sure to grasp the needle by the plastic cover and avoid touching the end of the syringe and the needle.
- Remove the plastic cap protecting the rubber stopper of the RAPTIVA vial. Open one alcohol prep pad package and wipe the rubber stopper with an alcohol prep pad. Do not touch the top of the vial after wiping.
- Remove the cap covering the prefilled syringe tip. Remove one of the 25-gauge needles from its package by grasping the needle by the plastic cover and without touching the end of the needle. Carefully place the capped 25-gauge needle onto the syringe tip. Twist needle to secure.
Mixing RAPTIVA
-
Remove the needle cap.
Do not touch the needle.
Keep the RAPTIVA vial upright on a firm surface, and slowly puncture the rubber stopper with the needle. Slowly push down on the syringe plunger to inject all of the 1.3 mL of sterile water onto the side wall of the vial to cause less foaming. Some foaming may happen; this is normal.
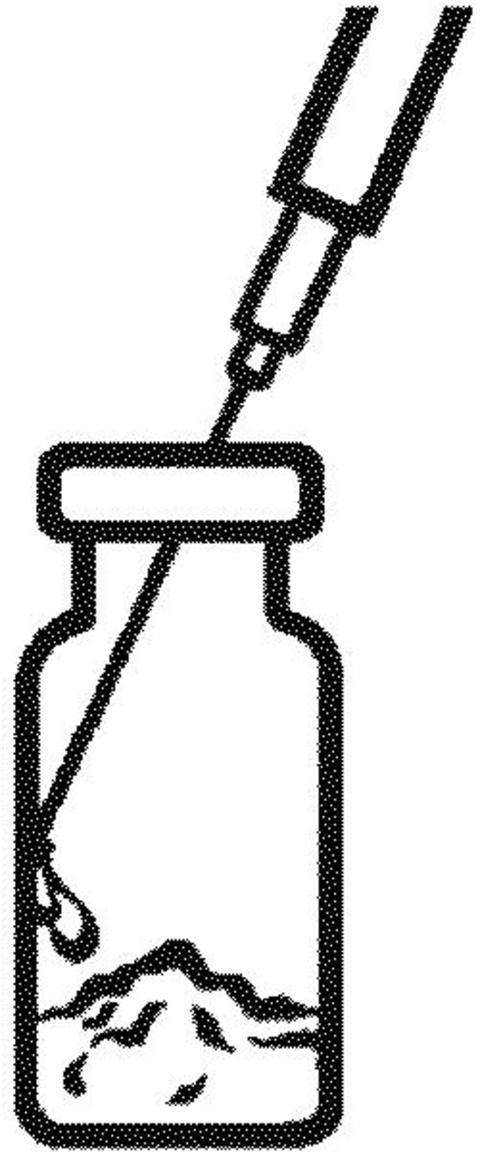
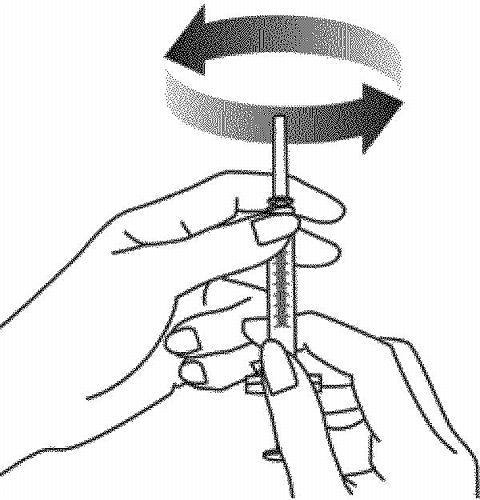
-
With the needle and syringe still in the vial stopper, gently swirl the vial to mix. Wait 5 minutes for the medicine to completely dissolve. To avoid excess foaming,
do not shake the vial.
The RAPTIVA solution should be clear to pale yellow.
Do not use the solution if it is discolored or cloudy or if particles (solid matter) are in the solution.
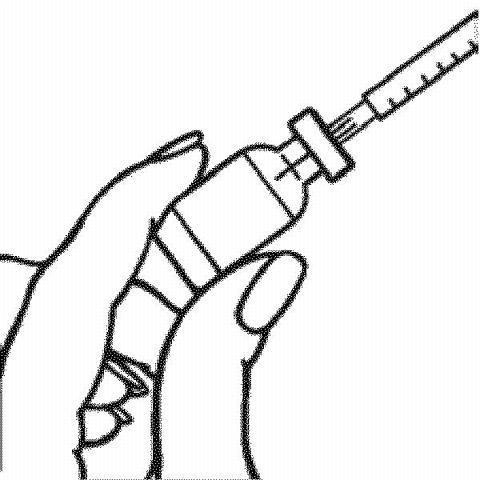
Preparing the RAPTIVA Dose for Injection
If you need more than one vial of RAPTIVA for the correct dose (dose amount is greater than 1.25 mL), repeat Steps 1-7 of this section using a second RAPTIVA blister tray, and divide your dose between two syringes.
- Turn the vial upside down, keeping the needle in the vial. (The needle will now be pointing upward.) Make sure the tip of the needle is covered all the way by the medicine in the vial. Pull back the syringe slightly if necessary. This will make it easier to get the medicine into the syringe.
-
Pull back on the plunger to fill the syringe. Withdraw the correct dose of medicine by reading the numbers on the syringe. Remove the syringe from the vial.
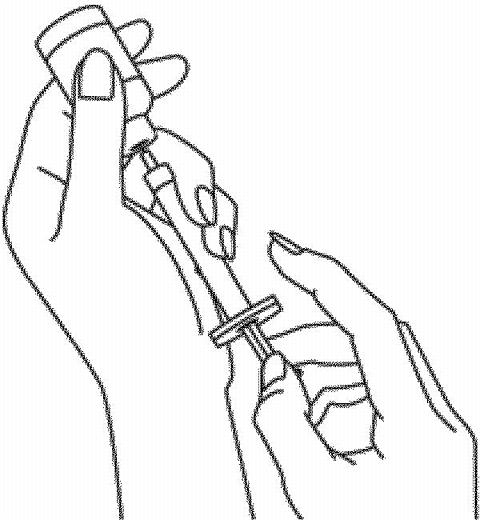
-
Slide the needle into the cap on a flat surface to pick up the needle cap. To lower the chance of a needlestick injury, do not touch the cap until it covers the needle all the way. Push the cap all the way down over the needle.

- Hold the syringe upright and tap the side of the syringe to let air bubbles rise to the top. Gently push in the plunger of the syringe to push the air bubbles out.
- After removing the bubbles, recheck the dose of medicine in the syringe. If necessary, push the plunger again to remove any amount of medicine beyond the line that indicates your dose. Make sure you have the right dose as instructed by your healthcare provider. Twist the capped needle off the syringe and discard it in a puncture-resistant container (see DISPOSAL OF THE SYRINGE, NEEDLES, AND SUPPLIES ). Never reuse a needle or syringe.
- Remove the other 25-gauge needle from its package by grasping the needle by the plastic cover and without touching the end of the needle. Carefully place the capped 25-gauge needle onto the syringe tip. Twist to secure. Put the syringe down while preparing your skin for injection.
Selecting and Preparing the Injection Site
-
Wash your hands well with soap and water.
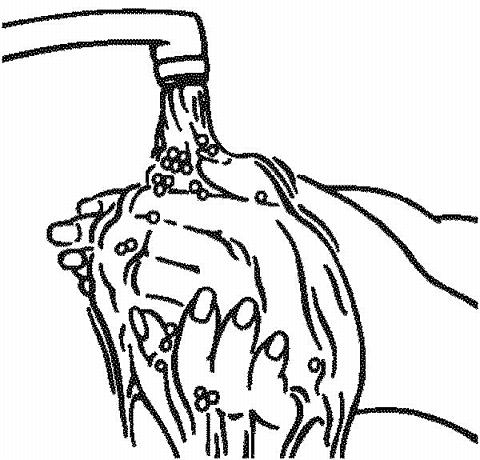
-
Choose an area of the body for the injection. Avoid, if possible, skin involved with psoriasis. Possible injection sites include the following:
- Outer area of the upper legs (thighs)
- Stomach area around the belly button
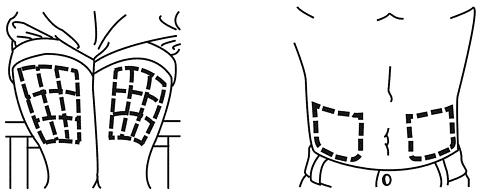
If someone else is giving you an injection, you can also use:
- Back of upper arms
- Buttocks
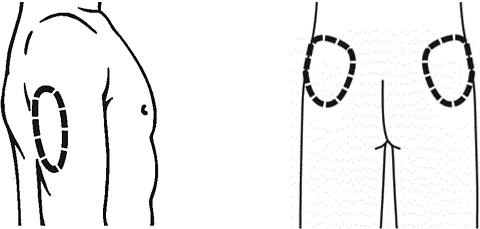
- It is important to change (rotate) the injection site each time you take RAPTIVA to lower your chances of soreness and redness at the injection site. Changing the injection site will also improve absorption of the medication. Repeat injections given in the same area should be at least 1 inch apart. Do not give an injection close to a vein that you can see under the surface of your skin.
- Wash the skin at the site of injection with soap and water. Let it air dry.
-
Cleanse the skin at the injection site with an alcohol prep pad using a circular motion. Let the area air dry all the way.
Do not touch this area again before giving the injection.
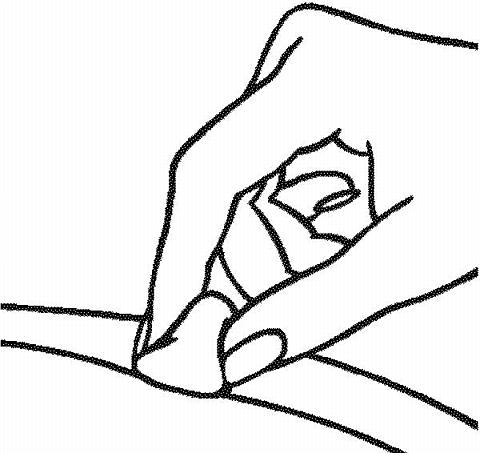
Giving the RAPTIVA Injection under the Skin
Your healthcare provider will teach you how to inject RAPTIVA. Do not inject RAPTIVA unless you have been taught the right way to give the injection.
-
Hold the syringe and remove the needle cover. Twisting the needle cover while pulling will help in the removal.
Do not
touch the needle or allow the needle to touch anything.
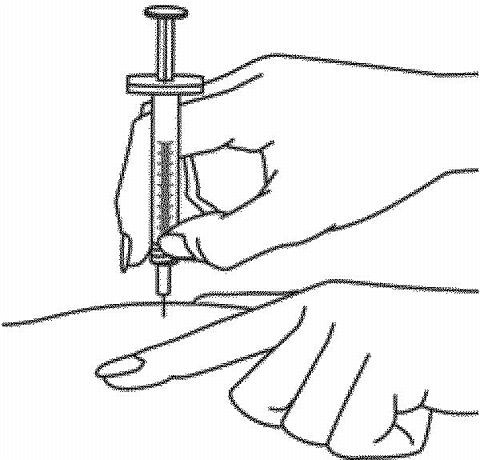
- Hold the syringe in the hand you use to inject yourself. Use your other hand to pinch a patch of skin at the clean injection site. Do not lay the syringe down or allow the needle to touch anything.
-
Hold the syringe firmly between your thumb and fingers so that you have steady control. Insert the needle straight down at a 90-degree angle. This is important to make sure the medicine is injected into fatty tissue.
- After the needle is inserted all the way into the skin, you can gently let go of the pinched skin. Be sure the needle stays in your skin. Slowly and smoothly push the plunger down into the syringe until it stops.
-
When all of the medicine has been injected, remove the needle and do not re-cap it. Discard the used syringe with the attached needle into a puncture resistant container (see
DISPOSAL OF THE SYRINGE, NEEDLES, AND SUPPLIES
).
Never reuse a needle or syringe.
Press a dry, sterile gauze (not provided) over the injection site. Do not use the alcohol prep pad. A small bandage may be put over the injection site.
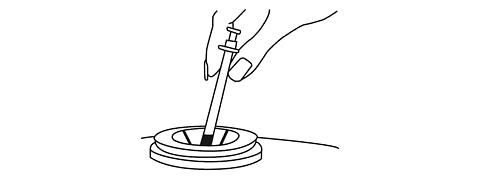
- If your dose amount is more than 1.25 mL, you will need to give a second injection. Choose the second injection site at least 1 inch from the first injection site.
DISPOSAL OF THE SYRINGE, NEEDLES, AND SUPPLIES
-
As stated earlier, place the used syringe with the attached needle in a puncture-resistant container, like a sharps container. You can buy a sharps container at your local pharmacy.
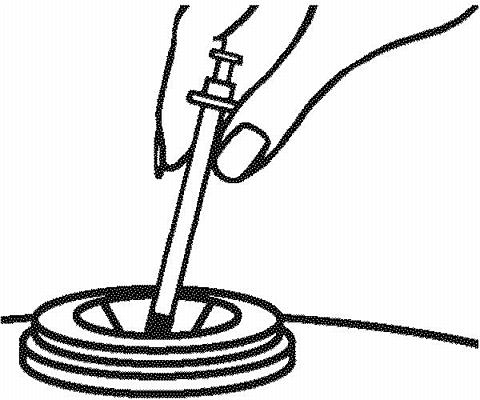
- Talk to your healthcare provider about how to properly dispose of a filled container of your used syringes and needles. There may be special local and state laws for disposing of used needles and syringes. Do not throw the filled container in the household trash and do not recycle.
- The needle cap, alcohol prep pads, and other used supplies can be thrown out with your regular trash.
- Always keep syringes, injection supplies, and disposal containers out of the reach of children.
- Do not reuse these single-use syringes or needles.
Rx Only
RAPTIVA® [efalizumab]
Manufactured by:
Genentech, Inc.
1 DNA Way
South San Francisco, CA 94080-4990
4826402
FDA Approval June 2005
Code Revision June 2005
©2005 Genentech, Inc.
Subscribe to the "News" RSS Feed 
TOP ۞