-
Zevalin Injection (Biogen Idec)
Kits for the Preparation of Indium-111 (In-111) Ibritumomab Tiuxetan (In-111 ZEVALIN) and Yttrium-90 (Y-90) Ibritumomab Tiuxetan (Y-90 ZEVALIN)
In-111 Ibritumomab Tiuxetan and Y-90 Ibritumomab Tiuxetan are components of the ZEVALIN therapeutic regimen (See Description ).
WARNINGS
Fatal Infusion Reactions: Deaths have occurred within 24 hours of Rituximab infusion, an essential component of the ZEVALIN therapeutic regimen. These fatalities were associated with an infusion reaction symptom complex that included hypoxia, pulmonary infiltrates, acute respiratory distress syndrome, myocardial infarction, ventricular fibrillation, or cardiogenic shock. Approximately 80% of fatal infusion reactions occurred in association with the first Rituximab infusion (See WARNINGS and ADVERSE REACTIONS ). Patients who develop severe infusion reactions should have Rituximab, In-111 ZEVALIN, and Y-90 ZEVALIN infusions discontinued and receive medical treatment.
Prolonged and Severe Cytopenias: Y-90 ZEVALIN administration results in severe and prolonged cytopenias in most patients. The ZEVALIN therapeutic regimen should not be administered to patients with >/= 25% lymphoma marrow involvement and/or impaired bone marrow reserve (See WARNINGS and ADVERSE REACTIONS ).
Severe Mucocutaneous Reactions: Severe mucocutaneous reactions, some with fatal outcome, have been reported in association with the ZEVALIN therapeutic regimen, which includes Rituximab, In-111 ZEVALIN and Y-90 ZEVALIN. (See WARNINGS and ADVERSE REACTIONS ).
Dosing
• The prescribed, measured, and administered dose of Y-90 ZEVALIN should not exceed the absolute maximum allowable dose of 32.0 mCi (1184 MBq).
• Y-90 ZEVALIN should not be administered to patients with altered biodistribution as determined by imaging with In-111 ZEVALIN.
In-111 ZEVALIN and Y-90 ZEVALIN are radiopharmaceuticals and should be used only by physicians and other professionals qualified by training and experienced in the safe use and handling of radionuclides.
DESCRIPTION
ZEVALIN®
ZEVALIN (Ibritumomab Tiuxetan) is the immunoconjugate resulting from a stable thiourea covalent bond between the monoclonal antibody Ibritumomab and the linker-chelator tiuxetan [N-[2-bis(carboxymethyl)amino]-3-(p-isothiocyanatophenyl)-propyl]-[N-[2-bis(carboxymethyl)amino]-2-(methyl)-ethyl]glycine. This linker-chelator provides a high affinity, conformationally restricted chelation site for Indium-111 or Yttrium-90. The approximate molecular weight of Ibritumomab Tiuxetan is 148 kD.
The antibody moiety of ZEVALIN is Ibritumomab, a murine IgG 1 kappa monoclonal antibody directed against the CD20 antigen, which is found on the surface of normal and malignant B lymphocytes. Ibritumomab is produced in Chinese hamster ovary cells and is composed of two murine gamma 1 heavy chains of 445 amino acids each and two kappa light chains of 213 amino acids each.
ZEVALIN Therapeutic Regimen
The ZEVALIN therapeutic regimen is administered in two steps: Step 1 includes one infusion of Rituximab preceding In-111 ZEVALIN. Step 2 follows Step 1 by seven to nine days and consists of a second infusion of Rituximab followed by Y-90 ZEVALIN.
ZEVALIN is supplied as two separate and distinctly labeled kits that contain all of the non-radioactive ingredients necessary to produce a single dose of In-111 ZEVALIN and a single dose of Y-90 ZEVALIN, both essential components of the ZEVALIN therapeutic regimen. Indium-111 chloride and Rituximab must be ordered separately from the ZEVALIN kit. Yttrium-90 Chloride Sterile Solution is supplied by MDS Nordion when the Y-90 ZEVALIN kit is ordered.
ZEVALIN Kits
Each of the two ZEVALIN kits contains four vials that are used to produce a single dose of either In-111 ZEVALIN or Y-90 ZEVALIN, as indicated on the outer container label:
- One (1) ZEVALIN vial containing 3.2 mg of Ibritumomab Tiuxetan in 2 mL of 0.9% sodium chloride solution; a sterile, pyrogen-free, clear, colorless solution that may contain translucent particles; no preservative present.
- One (1) 50 mM Sodium Acetate Vial containing 13.6 mg of sodium acetate trihydrate in 2 mL of Water for Injection; a sterile, pyrogen-free, clear, colorless solution; no preservative present.
- One (1) Formulation Buffer Vial containing 750 mg of Albumin (Human), 76 mg of sodium chloride, 28 mg of sodium phosphate dibasic dodecahydrate, 4 mg of pentetic acid, 2 mg of potassium phosphate monobasic and 2 mg of potassium chloride in 10 mL of Water for Injection adjusted to pH 7.1 with either sodium hydroxide or hydrochloric acid; a sterile, pyrogen-free, clear yellow to amber colored solution; no preservative present.
- One (1) empty Reaction Vial, sterile, pyrogen-free.
Physical/Radiochemical Characteristics of In-111
Indium-111 decays by electron capture, with a physical half-life of 67.3 hours (2.81 days). [1] The product of radioactive decay is nonradioactive cadmium-111. Radiation emission data for In-111 are summarized in Table 1.
Table 1.
Principal In-111 Radiation Emission DataRadiation Mean % per
DisintegrationMean
Energy (keV)Gamma-2 90.2 171.3 Gamma-3 94.0 245.4 External Radiation
The exposure rate constant for 37 MBq (1 mCi) of In-111 is 8.3 × 10 -4 C/kg/hr (3.2 R/hr) at 1 cm. Adequate shielding should be used with this gamma-emitter, in accordance with institutional good radiation safety practices.
To allow correction for physical decay of In-111, the fractions that remain at selected intervals before and after the time of calibration are shown in Table 2.
Table 2.
Physical Decay Chart: In-111
Half-life 2.81 Days (67.3 Hours)Calibration
Time (Hrs.)Fraction
Remaining-48 1.64 -42 1.54 -36 1.45 -24 1.28 -12 1.13 -6 1.06 0 1.00 6 0.94 12 0.88 24 0.78 36 0.69 42 0.65 48 0.61 Physical/Radiochemical Characteristics of Y-90
Yttrium-90 decays by emission of beta particles, with a physical half-life of 64.1 hours (2.67 days) [1] . The product of radioactive decay is non-radioactive zirconium-90. The range of beta particles in soft tissue ([khgr ] 90 ) is 5 mm. Radiation emission data for Y-90 are summarized in Table 3.
Table 3.
Principal Y-90 Radiation Emission DataRadiation Mean % per
DisintegrationMean
Energy (keV)Beta minus 100 750-935 External Radiation
The exposure rate for 37 MBq (1 mCi) of Y-90 is 8.3 × 10 -3 C/kg/hr (32 R/hr) at the mouth of an open Y-90 vial. Adequate shielding should be used with this beta-emitter, in accordance with institutional good radiation safety practices.
To allow correction for physical decay of Y-90, the fractions that remain at selected intervals before and after the time of calibration are shown in Table 4.
Table 4.
Physical Decay Chart: Y-90
Half-life 2.67 Days (64.1 Hours)Calibration
Time (Hrs.)Fraction
RemainingCalibration
Time (Hrs.)Fraction
Remaining-36 1.48 0 1.00 -24 1.30 1 0.99 -12 1.14 2 0.98 -8 1.09 3 0.97 -7 1.08 4 0.96 -6 1.07 5 0.95 -5 1.06 6 0.94 -4 1.04 7 0.93 -3 1.03 8 0.92 -2 1.02 12 0.88 -1 1.01 24 0.77 0 1.00 36 0.68 CLINICAL PHARMACOLOGY
General Pharmacology
Ibritumomab Tiuxetan binds specifically to the CD20 antigen (human B-lymphocyte-restricted differentiation antigen, Bp35). [2,3] The apparent affinity (K D ) of Ibritumomab Tiuxetan for the CD20 antigen ranges between approximately 14 to 18 nM. The CD20 antigen is expressed on pre-B and mature B lymphocytes and on > 90% of B-cell non-Hodgkin's lymphomas (NHL). [4,5] The CD20 antigen is not shed from the cell surface and does not internalize upon antibody binding. [6]
Mechanism of Action: The complementarity-determining regions of Ibritumomab bind to the CD20 antigen on B lymphocytes. Ibritumomab, like Rituximab, induces apoptosis in CD20+ B-cell lines in vitro . [6] The chelate tiuxetan, which tightly binds In-111 or Y-90, is covalently linked to the amino groups of exposed lysines and arginines contained within the antibody. The beta emission from Y-90 induces cellular damage by the formation of free radicals in the target and neighboring cells. [7]
Normal Human Tissue Cross-Reactivity: Ibritumomab Tiuxetan binding was observed in vitro on lymphoid cells of the bone marrow, lymph node, thymus, red and white pulp of the spleen, and lymphoid follicles of the tonsil, as well as lymphoid nodules of other organs such as the large and small intestines. Binding was not observed on the nonlymphoid tissues or gonadal tissues (see CLINICAL PHARMACOLOGY , Radiation Dosimetry )
Pharmacokinetics / Pharmacodynamics
Pharmacokinetic and biodistribution studies were performed using In-111 ZEVALIN (5 mCi [185 MBq] In-111, 1.6 mg Ibritumomab Tiuxetan). In an early study designed to assess the need for pre-administration of unlabeled antibody, only 18% of known sites of disease were imaged when In-111 ZEVALIN was administered without unlabeled Ibritumomab. When preceded by unlabeled Ibritumomab (1.0 mg/kg or 2.5 mg/kg), In-111 ZEVALIN detected 56% and 92% of known disease sites, respectively. These studies were conducted with a ZEVALIN therapeutic regimen that included unlabeled Ibritumomab.
In pharmacokinetic studies of patients receiving the ZEVALIN therapeutic regimen, the mean effective half-life for Y-90 activity in blood was 30 hours, and the mean area under the fraction of injected activity (FIA) vs. time curve in blood was 39 hours. Over 7 days, a median of 7.2% of the injected activity was excreted in urine.
In clinical studies, administration of the ZEVALIN therapeutic regimen resulted in sustained depletion of circulating B cells. At four weeks, the median number of circulating B cells was zero (range, 0-1084 cell/mm 3 ). B-cell recovery began at approximately 12 weeks following treatment, and the median level of B cells was within the normal range (32 to 341 cells/mm 3 ) by 9 months after treatment. Median serum levels of IgG and IgA remained within the normal range throughout the period of B-cell depletion. Median IgM serum levels dropped below normal (median 49 mg/dL, range 13-3990 mg/dL) after treatment and recovered to normal values by 6-month post therapy.
Radiation Dosimetry
Estimations of radiation-absorbed doses for In-111 ZEVALIN and Y-90 ZEVALIN were performed using sequential whole body images and the MIRDOSE 3 software program. [8,9] The estimated radiation absorbed doses to organs and marrow from a course of the ZEVALIN therapeutic regimen are summarized in Table 5. Absorbed dose estimates for the lower large intestine, upper large intestine, and small intestine have been modified from the standard MIRDOSE 3 output to account for the assumption that activity is within the intestine wall rather than the intestine contents.
Table 5.
Estimated Radiation Absorbed Doses From Y-90 ZEVALIN and In-111 ZEVALINY-90 ZEVALIN
mGy/MBqIn-111 ZEVALIN
mGy/MBqOrganMedian Range Median Range Spleen 19.4 1.8-20.0 0.9 0.2-1.8 Liver 14.8 2.9-8.1 0.7 0.4-1.1 Lower Large Intestinal Wall 14.7 3.1-8.2 0.4 0.2-0.6 Upper Large Intestinal Wall 13.6 2.0-6.7 0.3 0.2-0.6 Heart Wall 12.9 1.5-3.2 0.4 0.2-0.5 Lungs 12.0 1.2-3.4 0.2 0.2-0.4 Testes 11.5 1.0-4.3 0.1 0.1-0.3 Small Intestine 11.4 0.8-2.1 0.2 0.2-0.3 Red Marrow 21.3 0.6-1.8 0.2 0.1-0.2 Urinary Bladder Wall 30.9 0.7-1.3 0.2 0.1-0.2 Bone Surfaces 20.9 0.5-1.2 0.2 0.1-0.2 Total Body 30.5 0.4-0.7 0.1 0.1-0.2 Ovaries 30.4 0.3-0.5 0.2 0.2-0.2 Uterus 30.4 0.3-0.5 0.2 0.1-0.2 Adrenals 30.3 0.2-0.5 0.2 0.2-0.3 Brain 30.3 0.2-0.5 0.1 0.0-0.1 Breasts 30.3 0.2-0.5 0.1 0.1-0.1 Gallbladder Wall 30.3 0.2-0.5 0.3 0.2-0.4 Muscle 30.3 0.2-0.5 0.1 0.1-0.1 Pancreas 30.3 0.2-0.5 0.2 0.2-0.3 Skin 30.3 0.2-0.5 0.1 0.0-0.1 Stomach 30.3 0.2-0.5 0.2 0.1-0.2 Thymus 30.3 0.2-0.5 0.1 0.1-0.2 Thyroid 30.3 0.2-0.5 0.1 0.0-0.1 Kidneys 10.1 0.0-0.3 0.2 0.1-0.2 1 Organ region of interest2 Sacrum region of interest [10]3 Whole body region of interestCLINICAL STUDIES
The safety and efficacy of the ZEVALIN therapeutic regimen were evaluated in two multi-center trials enrolling a total of 197 subjects. The ZEVALIN therapeutic regimen was administered in two steps (see DOSAGE AND ADMINISTRATION ). The activity and toxicity of a variation of the ZEVALIN therapeutic regimen employing a reduced dose of Y-90 ZEVALIN was further defined in a third study enrolling a total of 30 patients who had mild thrombocytopenia (platelet count 100,000 to 149,000 cells/mm 3 ).
Study 1 was a single arm study of 54 patients with relapsed follicular lymphoma refractory to Rituximab treatment. Patients were considered refractory if their last prior treatment with Rituximab did not result in a complete or partial response, or if time to disease progression (TTP) was < 6 months. [11] The primary efficacy endpoint of the study was the overall response rate (ORR) using the International Workshop Response Criteria (IWRC). [12] Secondary efficacy endpoints included time to disease progression (TTP) and duration of response (DR). In a secondary analysis comparing objective response to the ZEVALIN therapeutic regimen with that observed with the most recent treatment with Rituximab, the median duration of response following the ZEVALIN therapeutic regimen was 6 vs. 4 months. Table 6 summarizes efficacy data from this study.
Study 2 was a randomized, controlled, multicenter study comparing the ZEVALIN therapeutic regimen to treatment with Rituximab. The trial was conducted in 143 patients with relapsed or refractory low-grade or follicular non-Hodgkin's lymphoma (NHL), or transformed B-cell NHL. A total of 73 patients received the ZEVALIN therapeutic regimen, and 70 patients received Rituximab given as an IV infusion at 375 mg/m 2 weekly times 4 doses. The primary efficacy endpoint of the study was to determine the ORR using the IWRC [12] (see Table 6). The ORR was significantly higher (80% vs. 56%, p = 0.002) [13] for patients treated with the ZEVALIN therapeutic regimen. The secondary endpoints, duration of response and time to progression, were not significantly different between the two treatment arms.
Table 6.
Summary of Efficacy Data 1Study 1 Study 2 ZEVALIN
therapeutic regimen
N = 54ZEVALIN
therapeutic regimen
N = 73Rituximab
N = 70Overall Response Rate (%)74 80 56 Complete Response Rate 2 (%)15 34 20 Median DR 3 , 4(Months)6.4 13.9 11.8 [Range 5 ][0.5-49.9 + ] [1.0-47.6 + ] [1.2-49.7 + ] Median TTP 3 , 6(Months)6.8 10.6 10.1 [Range 5 ][1.1-50.9 + ] [0.8-49.0 + ] [0.7-51.3 + ] 1 IWRC: International Workshop response criteria2 CRu and CR: Unconfirmed and confirm complete response3 Estimated with observed range4 Duration of response: interval from the onset of response to disease progression5 "+" indicates an ongoing response6 Time to Disease Progression: interval from the first infusion to disease progressionStudy 3 was a single arm study of 30 patients with relapsed or refractory low-grade, follicular, or transformed B-cell NHL who had mild thrombocytopenia (platelet count 100,000 to 149,000 cells/mm 3 ). Excluded from the study were patients with >/= 25% lymphoma marrow involvement and/or impaired bone marrow reserve. Patients were considered to have impaired bone marrow reserve if they had any of the following: prior myeloablative therapy with stem cell support; prior external beam radiation to > 25% of active marrow; a platelet count <100,000 cells/mm 3 ; or neutrophil count <1,500 cells/mm 3 . In this study, a modification of the ZEVALIN therapeutic regimen with a lower Y-90 ZEVALIN dose [Y-90 ZEVALIN at 0.3 mCi/kg (11.1 MBq/kg)] was used. Objective, durable clinical responses were observed [83% ORR (95% CI: 65-94%) [14] , 11.5 months median DR (range: 1-42.4+ months)] and resulted in a greater incidence of hematologic toxicity (see ADVERSE REACTIONS ) than in Studies 1 and 2.
INDICATIONS AND USAGE
ZEVALIN, as part of the ZEVALIN therapeutic regimen (see DOSAGE AND ADMINISTRATION ), is indicated for the treatment of patients with relapsed or refractory low-grade, follicular, or transformed B-cell non-Hodgkin's lymphoma, including patients with Rituximab refractory follicular non-Hodgkin's lymphoma. Determination of the effectiveness of the ZEVALIN therapeutic regimen in a relapsed or refractory patient population is based on overall response rates (see CLINICAL STUDIES ). The effects of the ZEVALIN therapeutic regimen on survival are not known.
CONTRAINDICATIONS
The ZEVALIN therapeutic regimen is contraindicated in patients with known Type I hypersensitivity or anaphylactic reactions to murine proteins or to any component of this product, including Rituximab, yttrium chloride, and indium chloride.
WARNINGS (SEE BOXED WARNING )
Altered Biodistribution: Y-90 ZEVALIN should not be administered to patients with altered biodistribution of In-111 ZEVALIN. For additional information regarding bio-distribution, see IMAGE ACQUISITION AND INTERPRETATION.
Severe Infusion Reactions (See PRECAUTIONS , Hypersensitivity): The ZEVALIN therapeutic regimen may cause severe, and potentially fatal, infusion reactions. These severe reactions typically occur during the first Rituximab infusion with time to onset of 30 to 120 minutes. Signs and symptoms of severe infusion reaction may include hypotension, angioedema, hypoxia, or bronchospasm, and may require interruption of Rituximab, In-111 ZEVALIN, or Y-90 ZEVALIN administration. The most severe manifestations and sequelae may include pulmonary infiltrates, acute respiratory distress syndrome, myocardial infarction, ventricular fibrillation, and cardiogenic shock. Because the ZEVALIN therapeutic regimen includes the use of Rituximab, see also prescribing information for RITUXAN (Rituximab).
Cytopenias (See ADVERSE REACTIONS, Hematologic Events ):
The most common severe adverse events reported with the ZEVALIN therapeutic regimen were thrombocytopenia (61% of patients with platelet counts <50,000 cells/mm 3 ) and neutropenia (57% of patients with absolute neutrophil count (ANC) <1,000 cells/mm 3 ) in patients with >/=150,000 platelets/mm 3 prior to treatment. Both incidences of severe thrombocytopenia and neutropenia increased to 78% and 74% for patients with mild thrombocytopenia at baseline (platelet count of 100,000 to 149,000 cells/mm 3 ). For all patients, the median time to nadir was 7-9 weeks and the median duration of cytopenias was 22-35 days. In <5% of cases, patients experienced severe cytopenia that extended beyond the prospectively defined protocol treatment period of 12 weeks following administration of the ZEVALIN therapeutic regimen. Some of these patients eventually recovered from cytopenia, while others experienced progressive disease, received further anti-cancer therapy, or died of their lymphoma without having recovered from cytopenia. The cytopenias may have influenced subsequent treatment decisions.
Hemorrhage, including fatal cerebral hemorrhage, and severe infections have occurred in a minority of patients in clinical studies. Careful monitoring for and management of cytopenias and their complications (e.g., febrile neutropenia, hemorrhage) for up to 3 months after use of the ZEVALIN therapeutic regimen are necessary. Caution should be exercised in treating patients with drugs that interfere with platelet function or coagulation following the ZEVALIN therapeutic regimen and patients receiving such agents should be closely monitored.
The ZEVALIN therapeutic regimen should not be administered to patients with >/= 25% lymphoma marrow involvement and/or impaired bone marrow reserve, e.g., prior myeloablative therapies; platelet count <100,000 cells/mm 3 ; neutrophil count <1,500 cells/mm 3 ; hypocellular bone marrow (</= 15% cellularity or marked reduction in bone marrow precursors); or to patients with a history of failed stem cell collection.
Severe Mucocutaneous Reactions (See BOXED WARNINGS and ADVERSE REACTIONS ): In postmarketing reports erythema multiforme, bullous dermatitis, exfoliative dermatitis, Stevens-Johnson syndrome and toxic epidermal necrolysis have been reported with patients who received the ZEVALIN therapeutic regimen, which includes Rituximab, In-111 ZEVALIN and Y-90 ZEVALIN. The onset of the reactions in the reported cases has ranged from days to months. Patients experiencing a severe mucocutaneous reaction should not receive any further components of the ZEVALIN therapeutic regimen and should seek prompt medical evaluation.
Secondary Malignancies: Out of 349 patients treated with the ZEVALIN therapeutic regimen, three cases of acute myelogenous leukemia and two cases of myelodysplastic syndrome have been reported following the ZEVALIN therapeutic regimen (see ADVERSE REACTIONS ).
Pregnancy Category D: Y-90 ZEVALIN can cause fetal harm when administered to a pregnant woman. There are no adequate and well-controlled studies in pregnant women. If this drug is used during pregnancy, or if the patient becomes pregnant while receiving this drug, the patient should be apprised of the potential hazard to the fetus. Women of childbearing potential should be advised to avoid becoming pregnant.
Creutzfeldt-Jakob disease (CJD): This product contains albumin, a derivative of human blood. Based on effective donor screening and product manufacturing processes, it carries an extremely remote risk for transmission of viral diseases. A theoretical risk for transmission of Creutzfeldt-Jakob disease (CJD) also is considered extremely remote. No cases of transmission of viral diseases or CJD have ever been identified for albumin.
PRECAUTIONS
The ZEVALIN therapeutic regimen is intended as a single course treatment. The safety and toxicity profile from multiple courses of the ZEVALIN therapeutic regimen or of other forms of therapeutic irradiation preceding, following, or in combination with the ZEVALIN therapeutic regimen have not been established.
Radionuclide Precautions: The contents of the ZEVALIN kit are not radioactive. However, during and after radiolabeling ZEVALIN with In-111 or Y-90, care should be taken to minimize radiation exposure to patients and to medical personnel, consistent with institutional good radiation safety practices and patient management procedures.
Hypersensitivity: Anaphylactic and other hypersensitivity reactions have been reported following the intravenous administration of proteins to patients. Medications for the treatment of hypersensitivity reactions, e.g., epinephrine, antihistamines and corticosteroids, should be available for immediate use in the event of an allergic reaction during administration of ZEVALIN. Patients who have received murine proteins should be screened for human anti-mouse antibodies (HAMA). Patients with evidence of HAMA have not been studied and may be at increased risk of allergic or serious hypersensitivity reactions during ZEVALIN therapeutic regimen administrations.
Immunization: The safety of immunization with live viral vaccines following the ZEVALIN therapeutic regimen has not been studied. Also, the ability of patients who received the ZEVALIN therapeutic regimen to generate a primary or anamnestic humoral response to any vaccine has not been studied.
Laboratory Monitoring: Complete blood counts (CBC) and platelet counts should be obtained weekly following the ZEVALIN therapeutic regimen and should continue until levels recover. CBC and platelet counts should be monitored more frequently in patients who develop severe cytopenia, or as clinically indicated.
Drug Interactions: No formal drug interaction studies have been performed with ZEVALIN. Due to the frequent occurrence of severe and prolonged thrombocytopenia, the potential benefits of medications which interfere with platelet function and/or anticoagulation should be weighed against the potential increased risks of bleeding and hemorrhage. Patients receiving medications that interfere with platelet function or coagulation should have more frequent laboratory monitoring for thrombocytopenia. In addition, the transfusion practices for such patients may need to be modified given the increased risk of bleeding.
Patients in clinical studies were prohibited from receiving growth factor treatment for 2 weeks prior to the ZEVALIN therapeutic regimen as well as for 2 weeks following completion of the regimen.
Carcinogenesis, Mutagenesis, Impairment of Fertility: No long-term animal studies have been performed to establish the carcinogenic or mutagenic potential of the ZEVALIN therapeutic regimen, or to determine its effects on fertility in males or females. However, radiation is a potential carcinogen and mutagen. The ZEVALIN therapeutic regimen results in a significant radiation dose to the testes. The radiation dose to the ovaries has not been established. There have been no studies to evaluate whether the ZEVALIN therapeutic regimen causes hypogonadism, premature menopause, azoospermia and/or mutagenic alterations to germ cells. There is a potential risk that the ZEVALIN therapeutic regimen could cause toxic effects on the male and female gonads. Effective contraceptive methods should be used during treatment and for up to 12 months following the ZEVALIN therapeutic regimen.
Pregnancy Category D: SEE WARNINGS.
Nursing Mothers: It is not known whether ZEVALIN is excreted in human milk. Because human IgG is excreted in human milk and the potential for ZEVALIN exposure in the infant is unknown, women should be advised to discontinue nursing and formula feeding should be substituted for breast feedings (see CLINICAL PHARMACOLOGY ).
Geriatric Use: Of 349 patients treated with the ZEVALIN therapeutic regimen in clinical studies, 38% (132 patients) were age 65 years and over, while 12% (41 patients) were age 75 years and over. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, but greater sensitivity of some older individuals cannot be ruled out.
Pediatric Use: The safety and effectiveness of the ZEVALIN therapeutic regimen in children have not been established.
ADVERSE REACTIONS
Safety data, except where indicated, are based upon 349 patients treated in 5 clinical studies with the ZEVALIN therapeutic regimen (see DOSAGE AND ADMINISTRATION ). Because the ZEVALIN therapeutic regimen includes the use of Rituximab, also see prescribing information for RITUXAN (Rituximab).
The most serious adverse reactions caused by the ZEVALIN therapeutic regimen include infections (predominantly bacterial in origin), allergic reactions (bronchospasm and angioedema), and hemorrhage while thrombocytopenic (resulting in deaths). In addition, patients who have received the ZEVALIN therapeutic regimen have developed myeloid malignancies and dysplasias. Fatal infusion reactions have occurred following the infusion of Rituximab. Prolonged and severe cytopenias, and in postmarketing reports, mucocutaneous reactions have been observed with the ZEVALIN therapeutic regimen. Please refer to the BOXED WARNINGS and WARNINGS sections for detailed descriptions of these reactions.
The most common toxicities reported were neutropenia, thrombocytopenia, anemia, gastrointestinal symptoms (nausea, vomiting, abdominal pain, and diarrhea), increased cough, dyspnea, dizziness, arthralgia, anorexia, anxiety, and ecchymosis. Hematologic toxicity was often severe and prolonged, whereas most non-hematologic toxicity was mild in severity. Table 7 lists adverse events that occurred in >/= 5% of patients. A more detailed description of the incidence and duration of hematologic toxicities, according to baseline platelet count (as an indicator of bone marrow reserve) is provided in Table 8, Hematologic Toxicity.
Table 7.
Incidence of Adverse Events in >/= 5% of Patients
Receiving the ZEVALIN therapeutic regimen **/*
(N = 349)All Grades
%Grade 3/4
%Any Adverse Event99 89 Body as a Whole80 12 Asthenia43 3 Infection29 5 Chills24 <1 Fever17 1 Abdominal Pain16 3 Pain13 1 Headache12 1 Throat Irritation10 0 Back Pain8 1 Flushing6 0 Cardiovascular System17 3 Hypotension6 1 Digestive System48 3 Nausea31 1 Vomiting12 0 Diarrhea9 <1 Anorexia8 0 Abdominal
enlargement5 0 Constipation5 0 Hemic and Lymphatic System98 86 Thrombocytopenia95 63 Neutropenia77 60 Anemia61 17 Ecchymosis7 <1 Metabolic and Nutritional Disorders23 3 Peripheral Edema8 1 Angioedema5 <1 Musculoskeletal System18 1 Arthralgia7 1 Myalgia7 <1 Nervous System27 2 Dizziness10 <1 Insomnia5 0 Respiratory System36 3 Dyspnea14 2 Increased Cough10 0 Rhinitis6 0 Bronchospasm5 0 Skin and Appendages28 1 Pruritus9 <1 Rash8 <1 Special Senses7 <1 Urogenital System6 <1 **/* Adverse events were followed for a period of 12 weeks following the first Rituximab infusion of the ZEVALIN therapeutic regimen
Note: All adverse events are included, regardless of relationshipThe following adverse events (except for those noted in Table 7) occurred in between 1 and 4% of patients during the treatment period: urticaria (4%), anxiety (4%), dyspepsia (4%), sweats (4%), petechia (3%), epistaxis (3%), allergic reaction (2%), and melena (2%).
Severe or life-threatening adverse events occurring in 1-5% of patients (except for those noted in Table 7) consisted of pancytopenia (2%), allergic reaction (1%), gastrointestinal hemorrhage (1%), melena (1%), tumor pain (1%), and apnea (1%). The following severe or life threatening events occurred in <1% of patients: angioedema, tachycardia, urticaria, arthritis, lung edema, pulmonary embolus, encephalopathy, hematemesis, subdural hematoma, and vaginal hemorrhage.
Hematologic Events: Hematologic toxicity was the most frequently observed adverse event in clinical trials. Table 8 presents the incidence and duration of severe hematologic toxicity for patients with normal baseline platelet count (>/= 150,000 cells/mm 3 ) treated with the ZEVALIN therapeutic regimen and patients with mild thrombocytopenia (platelet count 100,000 to 149,000 cells/mm 3 ) at baseline who were treated with a modified ZEVALIN therapeutic regimen that included a lower Y-90 ZEVALIN dose at 0.3 mCi/kg (11.1 MBq/kg).
Table 8.
Severe Hematologic ToxicityZEVALIN
therapeutic regimen
using 0.4 mCi/kg Y-90 Dose
(14.8 MBq/kg)Modified ZEVALIN
therapeutic regimen
using 0.3 mCi/kg Y-90 dose
(11.1 MBq/kg)ANCMedian nadir (cells/mm 3 )800 600 Per Patient Incidence
ANC <1000 cells/mm 357% 74% Per Patient Incidence
ANC <500 cells/mm 330% 35% Median Duration (Days) *
ANC <1000 cells/mm 322 29 PlateletsMedian nadir (cells/mm 3 )41,000 24,000 Per Patient Incidence
Platelets <50,000 cells/mm 361% 78% Per Patient Incidence
Platelets <10,000 cells/mm 310% 14% Median Duration (Days) #
Platelets <50,000 cells/mm 324 35 *Median duration of neutropenia for patients with ANC <1000 cells/mm 3 (Date from last laboratory value showing ANC >/=1000 cells/mm 3 to date of first laboratory value following nadir showing ANC >/=1000 cells/mm 3 , censored at initiation of next treatment or death)# Median duration of thrombocytopenia for patients with platelets <50,000 cells/mm 3 (Date from last laboratory value showing platelet count >/=50,000 cells/mm 3 to date of first laboratory value following nadir showing platelet count >/=50,000 cells/mm 3 , censored at initiation of next treatment or death)Median time to ANC nadir was 62 days, to platelet nadir was 53 days, and to hemoglobin nadir was 68 days. Information on growth factor use and platelet transfusions is based on 211 patients for whom data were collected. Filgrastim was given to 13% of patients and erythropoietin to 8%. Platelet transfusions were given to 22% of patients and red blood cell transfusions to 20%.
Infectious Events: During the first 3 months after initiating the ZEVALIN therapeutic regimen, 29% of patients developed infections. Three percent of patients developed serious infections comprising urinary tract infection, febrile neutropenia, sepsis, pneumonia, cellulitis, colitis, diarrhea, osteomyelitis, and upper respiratory tract infection. Life threatening infections were reported for 2% of patients that included sepsis, empyema, pneumonia, febrile neutropenia, fever, and biliary stent-associated cholangitis. During follow-up from 3 months to 4 years after the start of treatment with ZEVALIN, 6% of patients developed infections. Two percent of patients had serious infections comprising urinary tract infection, bacterial or viral pneumonia, febrile neutropenia, perihilar infiltrate, pericarditis, and intravenous drug-associated viral hepatitis. One percent of patients had life threatening infections that included bacterial pneumonia, respiratory disease, and sepsis.
Secondary Malignancies: A total of 2% of patients developed secondary malignancies following the ZEVALIN therapeutic regimen. One patient developed a Grade 1 meningioma, three developed acute myelogenous leukemia, and two developed a myelodysplastic syndrome. The onset of a second cancer was 8-34 months following the ZEVALIN therapeutic regimen and 4 to 14 years following the patients' diagnosis of NHL.
Immunogenicity: Of 211 patients who received the ZEVALIN therapeutic regimen in clinical trials and who were followed for 90 days, there were eight (3.8%) patients with evidence of human anti-mouse antibody (HAMA) (n=5) or human anti-chimeric antibody (HACA) (n=4) at any time during the course of the study. Two patients had low titers of HAMA prior to initiation of the ZEVALIN therapeutic regimen; one remained positive without an increase in titer while the other had a negative titer post-treatment. Three patients had evidence of HACA responses prior to initiation of the ZEVALIN therapeutic regimen; one had a marked increase in HACA titer while the other two had negative titers post-treatment. Of the three patients who had negative HAMA or HACA titers prior to the ZEVALIN therapeutic regimen, two developed HAMA in absence of HACA titers, and one had both HAMA and HACA positive titers post-treatment. Evidence of immunogenicity may be masked in patients who are lymphopenic. There has not been adequate evaluation of HAMA and HACA at delayed timepoints, concurrent with the recovery from lymphopenia at 6-12 months, to establish whether masking of the immunogenicity at early timepoints occurs. The data reflect the percentage of patients whose test results were considered positive for antibodies to Ibritumomab or Rituximab using kinetic enzyme immunoassays to Ibritumomab and Rituximab. The observed incidence of antibody positivity in an assay is highly dependent on the sensitivity and specificity of the assay and may be influenced by several factors including sample handling and concomitant medications. Comparisons of the incidence of HAMA/HACA to the ZEVALIN therapeutic regimen with the incidence of antibodies to other products may be misleading.
OVERDOSAGE
Doses as high as 0.52 mCi/kg (19.2 MBq/kg) of Y-90 ZEVALIN were administered in ZEVALIN therapeutic regimen clinical trials and severe hematological toxicities were observed. No fatalities or second organ injury resulting from overdosage administrations were documented. However, single doses up to 50 mCi (1850 MBq) of Y-90 ZEVALIN, and multiple doses of 20 mCi (740 MBq) followed by 40 mCi (1480 MBq) of Y-90 ZEVALIN were studied in a limited number of subjects. In these trials, some patients required autologous stem cell support to manage hematological toxicity.
DOSAGE AND ADMINISTRATION
The ZEVALIN therapeutic regimen is administered in two steps: Step 1 includes a single infusion of 250 mg/m 2 Rituximab (not included in the ZEVALIN kits) preceding a fixed dose of 5.0 mCi (1.6 mg total antibody dose) of In-111 ZEVALIN administered as a 10 minute IV push. Step 2 follows step 1 by seven to nine days and consists of a second infusion of 250 mg/m 2 of Rituximab prior to 0.4 mCi/kg of Y-90 ZEVALIN administered as a 10 minute IV push.
Rituximab Administration: NOTE THAT THE DOSE OF RITUXIMAB IS LOWER WHEN USED AS PART OF THE ZEVALIN THERAPEUTIC REGIMEN, AS COMPARED TO THE DOSE OF RITUXIMAB WHEN USED AS A SINGLE AGENT. DO NOT ADMINISTER RITUXIMAB AS AN INTRAVENOUS PUSH OR BOLUS. Hypersensitivity reactions may occur (see WARNINGS ). Premedication, consisting of acetaminophen and diphenhydramine, should be considered before each infusion of Rituximab.
ZEVALIN Therapeutic Regimen Dose Modification in Patients with Mild Thrombocytopenia: The Y-90 ZEVALIN dose should be reduced to 0.3 mCi/kg (11.1 MBq/kg) for patients with a baseline platelet count between 100,000 and 149,000 cells/mm 3 .
Two separate and distinctly-labeled kits are ordered for the preparation of a single dose each of In-111 ZEVALIN and Y-90 ZEVALIN. In-111 ZEVALIN and Y-90 ZEVALIN are radiopharmaceuticals and should be used only by physicians and other professionals qualified by training and experienced in the safe use and handling of radionuclides. Changing the ratio of any of the reactants in the radiolabeling process may adversely impact therapeutic results. In-111 ZEVALIN and Y-90 ZEVALIN should not be used in the absence of the Rituximab pre-dose.
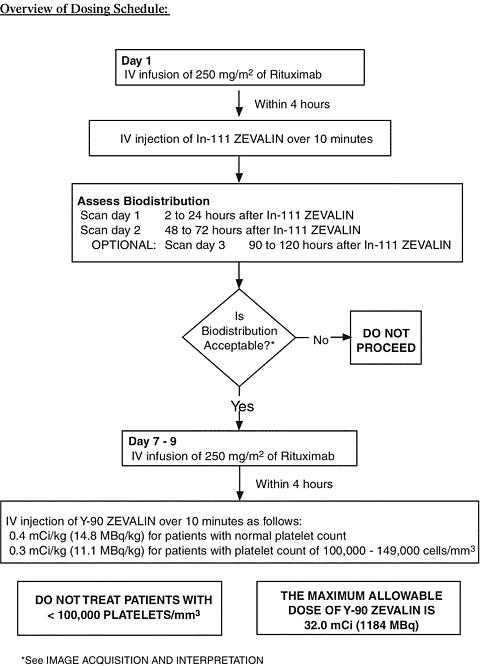
ZEVALIN Therapeutic Regimen Administration
Step 1:
First Rituximab Infusion: Rituximab at a dose of 250 mg/m 2 should be administered intravenously at an initial rate of 50 mg/hr. Rituximab should not be mixed or diluted with other drugs. If hypersensitivity or infusion-related events do not occur, escalate the infusion rate in 50 mg/hr increments every 30 minutes, to a maximum of 400 mg/hr. If hypersensitivity or an infusion-related event develops, the infusion should be temporarily slowed or interrupted (see WARNINGS ). The infusion can continue at one-half the previous rate upon improvement of patient symptoms.
In-111 ZEVALIN Injection: Within 4 hours following completion of the Rituximab dose, 5.0 mCi (1.6 mg total antibody dose) of In-111 ZEVALIN is injected intravenously (I.V.) over a period of 10 minutes. A 0.22 micrometer low-protein-binding filter should be in-line between the syringe and the infusion port prior to injection of In-111 ZEVALIN. After injection, the line should be flushed with at least 10 mL of normal saline.
Step 2:
Step 2 of the ZEVALIN therapeutic regimen is initiated seven to nine days following Step 1 administrations.
Second Rituximab Infusion: Rituximab at a dose of 250 mg/m 2 is administered I.V. at an initial rate of 100 mg/hr (50 mg/hr if infusion related events were documented during the first Rituximab administration) and increased by 100 mg/hr increments at 30 minute intervals, to a maximum of 400 mg/hr, as tolerated.
Y-90 ZEVALIN Injection:
Within 4 hours following completion of the Rituximab dose, Y-90 ZEVALIN at a dose of 0.4 mCi/kg (14.8 MBq/kg) actual body weight for patients with a platelet count >/=150,000 cells/mm 3 , and 0.3 mCi/kg (11.1 MBq/kg) actual body weight for patients with a platelet count of 100,000-149,000 cells/mm 3 is injected intravenously (I.V.) over a period of 10 minutes. A 0.22 micrometer low-protein-binding filter should be in-line between the syringe and the infusion port prior to injection of Y-90 ZEVALIN. After injection, the line should be flushed with at least 10 mL of normal saline. Precautions should be taken to avoid extravasation. A free flowing I.V. line should be established prior to Y-90 ZEVALIN injection. Close monitoring for evidence of extravasation during the injection of Y-90 ZEVALIN is required. If any signs or symptoms of extravasation have occurred, the infusion should be immediately terminated and restarted in another vein. The prescribed, measured, and administered dose of Y-90 ZEVALIN must not exceed the absolute maximum allowable dose of 32.0 mCi (1184 MBq), regardless of the patient's body weight. Do not give Y-90 ZEVALIN to patients with a platelet count <100,000/mm 3 (see WARNINGS ).
DIRECTIONS FOR PREPARATION OF RADIOLABELED ZEVALIN
-
PREPARATION OF THE IN-111 ZEVALIN DOSE
GENERAL:
Read all directions thoroughly and assemble all materials before starting the radiolabeling procedure. Important, significant differences exist in the preparation of the In-111 ZEVALIN dose and the Y-90 ZEVALIN dose.
The patient dose should be measured by a suitable radioactivity calibration system immediately prior to administration. The dose calibrator must be operated in accordance with the manufacturer's specifications and quality control for the measurement of In-111.
Proper aseptic technique and precautions for handling radioactive materials should be employed. Waterproof gloves should be utilized in the preparation and during the determination of radiochemical purity of In-111 ZEVALIN. Appropriate shielding should be used during radiolabeling, and use of a syringe shield is recommended during administration to the patient. The radiolabeling of ZEVALIN shall be done according to the following directions.
Required materials not supplied in the kit:
- Indium-111 Chloride Sterile Solution (In-111 Chloride) from Amersham Health, Inc. or Mallinckrodt, Inc.
- Three sterile 1 mL syringes
- One sterile 3 mL syringe
- Two sterile 10 mL syringes with 18-20 G needles
- Instant thin-layer chromatographic silica gel strips
- 0.9% sodium chloride aqueous solution for the chromatography solvent
- Developing chamber for chromatography
- Suitable radioactivity counting apparatus
- Filter, 0.22 micrometer, low-protein-binding (see DOSAGE AND ADMINISTRATION , Zevalin Therapeutic Regimen Administration)
- Vial and syringe shield
Method:
- Sterile, pyrogen-free In-111 chloride must be used for the preparation of In-111 ZEVALIN. The use of high purity In-111 chloride manufactured by Amersham Health, Inc. or Mallinckrodt, Inc. is required.
- Before radiolabeling, allow contents of the refrigerated carton to reach room temperature. Note: The ZEVALIN vial contains a protein solution that may develop translucent particulates. These particulates will be removed by filtration prior to administration.
- Clean the rubber stoppers of all of the vials in the kit and the In-111 chloride vial with a suitable alcohol swab and allow to air dry.
- Place the empty Reaction Vial in a suitable dispensing shield (pre-warmed to room temperature). To avoid the buildup of excessive pressure during the procedure, use a 10 mL syringe to withdraw 10 mL of air from the Reaction Vial.
-
Prior to initiating the radiolabeling reaction, determine the amount of each component needed according to the directions below:
- Calculate the volume of In-111 chloride that is equivalent to 5.5 mCi based on the activity concentration of the In-111 chloride stock.
- The volume of 50 mM sodium acetate solution needed is 1.2 times the volume of In-111 chloride solution determined in step 5.a., above. (The 50 mM sodium acetate is used to adjust the pH for the radiolabeling reaction.)
- Calculate the volume of Formulation Buffer needed to bring the Reaction Vial contents to a final volume of 10 mL. This is the volume of Formulation Buffer needed to protect the labeled product from radiolysis and to terminate the labeling reaction. For example, if volumes of 0.5 mL of In-111 chloride, 0.6 mL of sodium acetate and 1.0 mL of ZEVALIN were used, then the amount of formulation buffer would be 10-(0.5 + 0.6 + 1.0) = 7.9 mL.
- With a sterile 1 mL syringe, transfer the calculated volume of 50 mM of sodium acetate to the empty Reaction Vial. Coat the entire inner surface of the Reaction Vial by gentle inversion or rolling.
- Transfer 5.5 mCi of In-111 chloride to the Reaction Vial with a sterile 1 mL syringe. Mix the two solutions and coat the entire inner surface of the Reaction Vial by gentle inversion or rolling.
- With a sterile 3 mL syringe, transfer 1.0 mL of ZEVALIN (Ibritumomab Tiuxetan) to the Reaction Vial. Coat the entire surface of the Reaction Vial by gentle inversion or rolling. Do not shake or agitate the vial contents, since this will cause foaming and denaturation of the protein.
- Allow the labeling reaction to proceed at room temperature for 30 minutes. Allowing the labeling reaction to proceed for a longer or shorter time may result in inadequate labeling.
- Immediately after the 30-minute incubation period, using a sterile 10 mL syringe with a large bore needle (18 G - 20 G), transfer the calculated volume of Formulation Buffer from step 5.c. to the Reaction Vial. Gently add the Formulation Buffer down the side of the Reaction Vial. If necessary, to normalize air pressure, withdraw an equal volume of air. Coat the entire inner surface of the Reaction Vial by gentle inversion or rolling. Do not shake or agitate the vial contents. Avoid foaming.
- Using the supplied labels, record the patient identification, the date and time of preparation, the total activity and volume, and the date and time of expiration, and affix these labels to the reaction vial and shielded reaction vial container.
- Calculate the volume required for an In-111 ZEVALIN dose of 5 mCi. Withdraw the required volume from the Reaction Vial contents into a sterile 10 mL syringe with a large bore needle (18 G - 20 G). Assay the syringe and contents in a dose calibrator. The syringe should contain the dose of In-111 ZEVALIN to be administered to the patient. Using the supplied labels, record the patient identification, the date and time of preparation, the total activity and volume added, and the date and time of expiration, and affix these labels to the syringe and shielded unit dose container.
- Determine Radiochemical purity. See Section C: Procedure for Determining Radiochemical Purity Section that follows DIRECTIONS FOR PREPARATION OF THE Y-90 ZEVALIN DOSE.
- Indium-111 ZEVALIN should be stored at 2-8°C (36-46°F) until use and administered within 12 hours of radiolabeling.
- See DOSAGE AND ADMINISTRATION: ZEVALIN Therapeutic Regimen Administration: Step 1
- Discard vials, needles and syringes in accordance with local, state, and federal regulations governing radioactive and biohazardous waste.
-
PREPARATION OF THE Y-90 ZEVALIN DOSE
GENERAL:
Read all directions thoroughly and assemble all materials before starting the radiolabeling procedure. Important, significant differences exist in the preparation of the In-111 ZEVALIN dose and the Y-90 ZEVALIN dose.
The patient dose should be measured by a suitable radioactivity calibration system immediately prior to administration. The dose calibrator must be operated in accordance with the manufacturer's specifications and quality control for the measurement of Y-90.
Proper aseptic technique and precautions for handling radioactive materials should be employed. Waterproof gloves should be utilized in the preparation and during the determination of radiochemical purity of Y-90 ZEVALIN. Appropriate shielding should be used during radiolabeling, and use of a syringe shield is recommended during administration to the patient. The radiolabeling of ZEVALIN shall be done according to the following directions.
Required materials not supplied in the kit:
- Yttrium-90 Chloride Sterile Solution from MDS Nordion (shipped directly from MDS Nordion upon placement of an order for the Y-90 ZEVALIN kit)
- Three sterile 1 mL syringes
- One sterile 3 mL syringe
- Two sterile 10 mL syringes with 18-20 G needles
- Instant thin-layer chromatographic silica gel strips (ITLC-SG)
- 0.9% sodium chloride aqueous solution for the chromatography solvent
- Suitable radioactivity counting apparatus
- Developing chamber for chromatography
- Filter, 0.22 micrometer, low-protein-binding (see DOSAGE AND ADMINISTRATION , ZEVALIN Therapeutic Regimen Administration)
- Vial and syringe shield
Method:
- Sterile, pyrogen-free Y-90 chloride must be used for the preparation of Y-90 ZEVALIN. The use of high purity Y-90 chloride manufactured by MDS Nordion is required.
- Before radiolabeling, allow the contents of the refrigerated carton to reach room temperature. Note: The ZEVALIN vial contains a protein solution that may develop translucent particulates. These particulates will be removed by filtration prior to administration.
- Clean the rubber stoppers of all of the vials in the kit and the Y-90 chloride vial with a suitable alcohol swab and allow to air dry.
- Place the empty Reaction Vial in a suitable dispensing shield (pre-warmed to room temperature). To avoid the buildup of excessive pressure during the procedure, use a 10 mL syringe to withdraw 10 mL of air from the Reaction Vial.
-
Prior to initiating the radiolabeling reaction, determine the amount of each component needed according to the directions below:
- Calculate the volume of Y-90 chloride that is equivalent to 40 mCi based on the activity concentration of the Y-90 chloride stock.
- The volume of 50 mM sodium acetate solution needed is 1.2 times the volume of Y-90 chloride solution determined in step 5.a., above. (The 50 mM sodium acetate is used to adjust the pH for the radiolabeling reaction.)
- Calculate the volume of Formulation Buffer needed to bring the Reaction Vial contents to a final volume of 10 mL. This is the volume of Formulation Buffer needed to protect the labeled product from radiolysis and to terminate the labeling reaction. For example if the volumes were 0.5 mL of Y-90 chloride, 0.6 mL of sodium acetate and 1.3 mL of ZEVALIN, then the amount of formulation buffer would be 10 - (0.5 + 0.6 + 1.3) = 7.6 mL.
- With a sterile 1 mL syringe, transfer the calculated volume of 50 mM sodium acetate to the empty Reaction Vial. Coat the entire inner surface of the Reaction Vial by gentle inversion or rolling.
- Transfer 40 mCi of Y-90 chloride to the Reaction Vial with a sterile 1 mL syringe. Mix the two solutions and coat the entire inner surface of the Reaction Vial by gentle inversion or rolling.
- With a sterile 3 mL syringe, transfer 1.3 mL of ZEVALIN (Ibritumomab Tiuxetan) to the Reaction Vial. Coat the entire surface of the Reaction Vial by gentle inversion or rolling. Do not shake or agitate the vial contents, since this will cause foaming and denaturation of the protein.
- Allow the labeling reaction to proceed at room temperature for 5 minutes. Allowing the labeling reaction to proceed for a longer or shorter time may result in inadequate labeling.
- Immediately after the 5-minute incubation period, using a sterile 10 mL syringe with a large bore needle (18 G - 20 G), transfer the calculated volume of Formulation Buffer from step 5.c. to the Reaction Vial, terminating incubation. Gently add the Formulation Buffer down the side of the Reaction Vial. If necessary to normalize air pressure, withdraw an equal volume of air. Coat the entire inner surface of the Reaction Vial by gentle inversion or rolling. Do not shake or agitate the vial contents. Avoid foaming.
- Using the supplied labels, record the patient identification, the date and time of preparation, the total activity and volume, and the date and time of expiration and affix these labels to the reaction vial and shielded reaction vial container.
- Calculate the volume required for a Y-90 ZEVALIN dose of 0.4 mCi/kg (14.8 MBq/kg) actual body weight for patients with normal platelet count, and 0.3 mCi/kg (11.1 MBq/kg) actual body weight for patients with platelet count of 100,000 - 149,000 cells/mm 3 . The prescribed, measured, and administered dose of Y-90 ZEVALIN must not exceed the absolute maximum allowable dose of 32.0 mCi (1184 MBq), regardless of the patient's body weight. Withdraw the required volume from the Reaction Vial contents into a sterile 10 mL syringe with a large bore needle (18 G - 20 G). Assay the syringe and contents in a dose calibrator. The dose calibrator must be operated in accordance with the manufacturer's specifications and quality control for the measurement of Y-90. The syringe should contain the dose of Y-90 ZEVALIN to be administered to the patient, and should be within 10% of the actual prescribed dose of Y-90 ZEVALIN, not to exceed a maximum dose of 32.0 mCi. Do not exceed ± 10% of the prescribed dose. Using the supplied labels, record the patient identification, the date and time of preparation, the total activity and volume added, and the date and time of expiration and affix these labels to the syringe and shielded unit dose container.
- Determine Radiochemical Purity. See Section C: Procedure for Determining Radiochemical Purity Section that follows these DIRECTIONS FOR PREPARATION OF THE Y-90 ZEVALIN DOSE.
- Yttrium-90 ZEVALIN should be stored at 2-8°C (36-46°F) until use and administered within 8 hours of radiolabeling.
- See DOSAGE AND ADMINISTRATION: ZEVALIN Therapeutic Regimen Administration: Step 2 .
-
Discard vials, needles and syringes in accordance with local, state, and federal regulations governing radioactive and biohazardous waste.
Yttrium-90 ZEVALIN is suitable for administration on an outpatient basis. Beyond the use of vial and syringe shields for preparation and injection, no special shielding is necessary.
-
PROCEDURE FOR DETERMINING RADIOCHEMICAL PURITY (RCP)
The following procedure should be used for both In-111 ZEVALIN and Y-90 ZEVALIN:
- At room temperature, place a small drop of either In-111 ZEVALIN or Y-90 ZEVALIN at the origin of an ITLC-SG strip.
- Place the ITLC-SG strip into a chromatography chamber with the origin at the bottom and the solvent front at the top. Allow the solvent (0.9% NaCl) to migrate at least 5 cm from the bottom of the strip. Remove the strip from the chamber and cut the strip in half. Count each half of the ITLC-SG strip for one minute (CPM) with a suitable counting apparatus.
-
Calculate the percent RCP as follows:
% RCP = CPM bottom half
CPM bottom half + CPM top half× 100
- If the radiochemical purity is <95%, the ITLC procedure should be repeated. If repeat testing confirms that radiochemical purity is <95%, the preparation should not be administered.
IMAGE ACQUISITION AND INTERPRETATION
The biodistribution of In-111 ZEVALIN should be assessed by a visual evaluation of whole body planar view anterior and posterior gamma images at 2 - 24 hours and 48 - 72 hours after injection. To resolve ambiguities, a third image at 90 - 120 hours may be necessary. Images should be acquired using a large field of view gamma camera equipped with a medium energy collimator. Whole body anterior/posterior planar images should be acquired using a large field-of-view gamma camera and medium energy collimators. Suggested gamma camera settings: 256 × 1024 matrix; dual energy photopeaks set at 172 and 247 keV; 15% symmetric window; scan speed of 10 cm/min for the 2-24 hour scan, 7-10 cm/min for the 48-72 hour scan and 5 cm/min for the optional 90-120 hour scan.
EXPECTED BIODISTRIBUTION
Visual inspection of gamma images of expected biodistribution reveal the following:
- On Scan 1 (2-24 hours), activity in the blood pool areas (heart, abdomen, neck, and extremities) is detectable and decreases on Scan 2 (48-72 hours). There is variability within patients in the visualization of the blood pool especially when images are performed late in the time window of Scan 1 and in an occasional patient, blood pool may not be visible late in the time window of Scan 1.
- Moderately high to high uptake in normal liver and spleen on Scans 1 and 2.
- Moderately low or very low uptake in normal kidneys, urinary bladder, and normal (uninvolved) bowel on Scans 1 and 2.
- Localization to lymphoid aggregates in the bowel wall has been reported.
Tumor uptake may be visualized in soft tissue as areas of increased intensity, and tumor-bearing areas in normal organs may be seen as areas of increased or decreased intensity. Tumor visualization on the In-111 Zevalin scan is not required for Y-90 Zevalin therapy.
ALTERED BIODISTRIBUTION
The criteria for altered biodistribution is met if any one of the following is detected on visual inspection of gamma images:
- Rapid clearance of the radioimmunoconjugate from the blood pool with liver, spleen, and/or bone marrow uptake in Scan 1.
-
Increased uptake in normal organs (not involved by tumor) such as:
- Diffuse uptake in normal lung more intense than the cardiac blood pool on Scan 1, or more intense than the liver Scan 2.
- Kidneys with greater intensity than the liver on the posterior view on Scan 2. Fixed areas (unchanged with time) of uptake in the normal bowel greater than uptake in the liver on Scan 2.
- In less than 0.5% of patients receiving In-111 ZEVALIN, prominent bone marrow uptake was observed, characterized by clear visualization of the long bones and ribs on Scan 1.
If a visual inspection of the gamma images reveals an altered biodistribution, the patient should not proceed to the Y-90 ZEVALIN dose. The safety and efficacy of the administration of Y-90 ZEVALIN in patients with prominent marrow uptake is not known. Possible causes of prominent bone marrow uptake, such as bone marrow involvement by lymphoma, increased marrow activity due to recent hematopoietic growth factor administration, and increased reticuloendothelial uptake in patients with HAMA and HACA, should be considered. Re-assessment of biodistribution after correction of underlying factors should be performed. Y-90 ZEVALIN should not be administered to patients with persistently prominent marrow uptake on the repeat biodistribution scans.
During ZEVALIN clinical development, individual tumor radiation absorbed dose estimates as high as 778 cGy/mCi have been reported. Although solid organ toxicity has not been directly attributed to radiation from adjacent tumors, careful consideration should be applied before proceeding with treatment in patients with very high tumor uptake next to critical organs or structures.
HOW SUPPLIED
The In-111 ZEVALIN kit provides for the radiolabeling of Ibritumomab Tiuxetan with In-111. The Y-90 ZEVALIN kit provides for the radiolabeling of Ibritumomab Tiuxetan with Y-90.
The kit for the preparation of a single dose of In-111 ZEVALIN includes four vials: one ZEVALIN vial containing 3.2 mg of Ibritumomab Tiuxetan in 2 mL of 0.9% sodium chloride solution; one 50 mM Sodium Acetate vial; one Formulation Buffer vial; one empty Reaction Vial and four identification labels.
The kit for the preparation of a single dose of Y-90 ZEVALIN includes four vials: one ZEVALIN vial containing 3.2 mg of Ibritumomab Tiuxetan in 2 mL of 0.9% sodium chloride solution; one 50 mM Sodium Acetate vial; one Formulation Buffer vial; one empty Reaction Vial and four identification labels.
The contents of all vials are sterile, pyrogen-free and contain no preservatives.
The Indium-111 Chloride Sterile Solution (In-111 Chloride) must be ordered separately from either Amersham Health, Inc. or Mallinckrodt, Inc. at the time the In-111 ZEVALIN kit is ordered. The Yttrium-90 Chloride Sterile Solution will be shipped directly from MDS Nordion upon placement of an order for the Y-90 ZEVALIN kit.
Storage
Store at 2-8°C (36-46°F). Do not freeze.
REFERENCES
- Kocher DC. Radioactive Decay Data Tables - A Handbook of Decay Data for Application to Radiation Dosimetry and Radiological Assessments: U.S. Department of Energy 1981; DOE/TIC-11026.
- Valentine MA, Meier KE, Rossie S, Clark EA. Phosphorylation of the CD20 phosphoprotein in resting B lymphocytes. Regulation by protein kinase C. J Biol Chem 1989;264(19):11282-7.
- Einfeld D, Brown J, Valentine M, Clark E, Ledbetter J. Molecular cloning of the human B cell CD20 receptor predicts a hydrophobic protein with multiple transmembrane domains. EMBO J 1988;7(3):711-7.
- Anderson KC, Bates MP, Slaughenhoupt BL, Pinkus GS, Schlossman SF, Nadler LM. Expression of human B cell-associated antigens on leukemias and lymphomas: a model of human B cell differentiation. Blood 1984;63(6):1424-33.
- Tedder T, Boyd A, Freedman A, Nadler L, Schlossman S. The B cell surface molecule B1 is functionally linked with B cell activation and differentiation. J Immunol 1985;135(2):973-9.
- Press O, Appelbaum F, Ledbetter J, Martin P, Zarling J, Kidd P, et al. Monoclonal antibody 1F5 (anti-CD20) serotherapy of human B-cell lymphomas. Blood 1987;69(2):584-91.
- Chakrabarti MC, Le N, Paik CH, De Graff WG, Carrasquillo JA. Prevention of radiolysis of monoclonal antibody during labeling. J Nucl Med 1996;37(8):1384-8.
- Wiseman GA, White CA, Stabin M, Dunn WL, Erwin W, Dahlbom M, et al. Phase I/II 90Y ZevalinTM (90 yttrium ibritumomab tiuxetan, IDEC-Y2B8) radioimmunotherapy dosimetry results in relapsed or refractory non-Hodgkin's lymphoma. Eur J Nucl Med 2000;27(7):766-77.
- Wiseman GA, White CA, Sparks RB, Erwin WD, Podoloff DA, Lamonica DA, et al. Biodistribution and dosimetry results from a phase III prospectively randomized controlled trial of Zevalin radioimmunotherapy for low-grade, follicular, or transformed B-cell non-Hodgkin's lymphoma. Crit Rev Oncol Hematol 2001;39(1-2):181-94.
- Siegel J, Lee R, Pawlyk D, Horowitz J, Sharkey R, Goldenberg D. Sacral scintigraphy for bone marrow dosimetry in radioimmunotherapy. Int J Appl Radiat Instr 1989;16(Part B):553-9.
- Witzig TE, Flinn IW, Gordon LI, Emmanouilides C, Czuczman MS,Saleh MN, et al. Treatment with ibritumomab tiuxetan radioimmunotherapy in patients with rituximab-refractory follicular non-Hodgkin's Lymphoma. J Clin Oncol 2002;20(15):3262-9.
- Cheson BD, Horning SJ, Coiffier B, Shipp MA, Fisher RI, Connors JM, et al. Report of an international workshop to standardize response criteria for non-Hodgkin's lymphoma. J Clin Oncol 1999;17(4):1244-53.
- Witzig TE, Gordon LI, Cabanillas F, Czuczman MS, Emmanouilides C, Joyce R, et al. Randomized controlled trial of yttrium-90-labeled ibritumomab tiuxetan radioimmunotherapy versus rituximab immunotherapy for patients with relapsed or refractory low-grade, follicular, or transformed B-cell non-hodgkins lymphoma. J Clin Oncol. 2002;20(10):2453-63.
- Wiseman GA, Gordon LI, Multani PS, Witzig TE, Spies S, Bartlett NL, et al. Ibritumomab tiuxetan radioimmunotherapy for relapsed or refractory non-hodgkins lymphoma patients with mild thrombocytopenia: a phase II multicenter trial. Blood 2002; 99(12):4336-4342.
Rx Only
In-111 ZEVALIN kit, NDC 64406-104-04
Y-90 ZEVALIN kit, NDC 64406-103-03
© 2002, 2005 Biogen Idec Inc.
14 Cambridge Center
Cambridge, MA 02142
U.S. License Number 1697
Protected by one or more U.S. Patents.
Issue date: April 2005
Subscribe to the "News" RSS Feed 
TOP ۞