-
Nipent for Injection (Supergen)
WARNING
NIPENT should be administered under the supervision of a physician qualified and experienced in the use of cancer chemotherapeutic agents. The use of higher doses than those specified (see DOSAGE AND ADMINISTRATION ) is not recommended. Dose-limiting severe renal, liver, pulmonary, and CNS toxicities occurred in Phase 1 studies that used NIPENT at higher doses (20-50 mg/m 2 in divided doses over 5 days) than recommended.
In a clinical investigation in patients with refractory chronic lymphocytic leukemia using NIPENT at the recommended dose in combination with fludarabine phosphate, 4 of 6 patients entered in the study had severe or fatal pulmonary toxicity. The use of NIPENT in combination with fludarabine phosphate is not recommended.
DESCRIPTION
NIPENT® (pentostatin for injection) is supplied as a sterile, apyrogenic, lyophilized powder in single-dose vials for intravenous administration. Each vial contains 10 mg of pentostain and 50 mg of Mannitol, USP. The pH of the final product is maintained between 7.0 and 8.5 by addition of sodium hydroxide or hydrochloric acid.
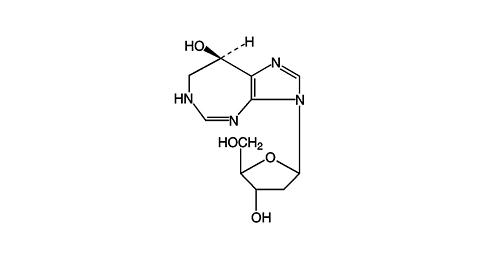
Pentostatin, also known as 2'-deoxycoformycin (DCF), is a potent inhibitor of the enzyme adenosine deaminase and is isolated from fermentation cultures of Streptomyces antibioticus. Pentostatin is known chemically as (R)-3-(2-deoxy-(beta)-D- erythro -pentofuranosyl)-3,6,7,8-tetrahydroimidazo[4,5- d ] [1,3]diazepin-8-ol with a molecular formula of C 11 H 16 N 4 O 4 and a molecular weight of 268.27.
Pentostain is a white to off-white solid, freely soluble in distilled water.
The molecular structure of pentostatin is:
CLINICAL PHARMACOLOGY
Mechanism of Action
Pentostatin is a potent transition state inhibitor of the enzyme adenosine deaminase (ADA). The greatest activity of ADA is found in cells of the lymphoid system with T-cells having higher activity than B-cells and T-cell malignancies higher ADA activity than B-cell malignancies. Pentostatin inhibition of ADA, particularly in the presence of adenosine or deoxyadenosine, leads to cytotoxicity, and this is believed to be due to elevated intracellular levels of dATP which can block DNA synthesis through inhibition of ribonucleotide reductase. Pentostatin can also inhibit RNA synthesis as well as cause increased DNA damage. In addition to elevated dATP, these mechanisms may also contribute to the overall cytotoxic effect of pentostatin. The precise mechanism of pentostain's antitumor effect, however, in hairy cell leukemia is not known.
Pharmacokinetics/Drug Metabolism
A tissue distribution and whole-body autoradiography study in the rat revealed that radioactivity concentrations were highest in the kidneys with very little central nervous system penetration.
In man, following a single dose of 4 mg/m 2 of pentostain infused over 5 minutes, the distribution half-life was 11 minutes, the mean terminal half-life was 5.7 hours, the mean plasma clearance was 68 mL/min/m 2 , and approximately 90% of the dose was excreted in the urine as unchanged pentostatin and/or metabolites as measured by adenosine deaminase inhibitory activity. The plasma protein binding of pentostain is low, approximately 4%.
A positive correlation was observed between pentostatin clearance and creatinine clearance (CrCl) in patients with creatinine clearance values ranging from 60 mL/min to 130 mL/min. 1 Pentostatin half-life in patients with renal impairment (CrCl <50 mL/min, n=2) was 18 hours, which was much longer than that observed in patients with normal renal function (CrCl >60 mL/min, n=14), about 6 hours.
CLINICAL STUDIES
The following table provides efficacy results for 4 groups (columns) of patients with hairy cell leukemia: patients who initially received NIPENT, patients who initially received alpha-interferon (IFN), and 2 different groups of patients who received NIPENT after proving to be refractory to, or intolerant of IFN therapy. The first 2 groups represent treatment results from the SWOG 8691 study, a large multicenter study comparing NIPENT and IFN in untreated (frontline) patients with confirmed hairy cell leukemia. The third group represents evaluable patients from the SWOG study who crossed over to NIPENT after initially receiving IFN. The fourth group, labeled NCI Phase 2 studies, displays pooled results of 2 noncomparative studies (MD Anderson and CALGB), in which NIPENT was used to treat patients with confirmed IFN-refractory disease.
In the SWOG 8691 study, NIPENT was administered at a dose of 4 mg/m 2 every 2 weeks. After 6 months of treatment, patients were evaluated for response. If a complete response was achieved, 2 additional doses of NIPENT were administered and then discontinued. If a partial response was achieved. NIPENT was continued for up to an additional 6 months. NIPENT was discontinued for stable disease after 6 months or progressive disease after 2 months of therapy. IFN was administered 3 million units subcutaneously 3 times per week. Patients who achieved a complete or partial response after 6 months of treatment continued on IFN for another 6 months. IFN was discontinued if patients did not achieve a complete or partial response after 6 months of initial treatment or progressed after 2 months. This study allowed crossover of patients intolerant of, or refractory to, initial treatment.
Interferon-refractory patients enrolled into the MD Anderson study received NIPENT at a dose of 4 mg/m 2 every other week for 3 months and responding patients received 3 additional months. CALGB patients received 4 mg/m 2 of NIPENT every other week for 3 months and responding patients were treated monthly for up to 9 additional months. Almost all patients had a PS of 0 to 2 in the Phase 2 and 3 studies.
For each study, a complete response (CR) required clearing of the peripheral blood and bone marrow of all hairy cells, normalization of organomegaly and lymphadenopathy by physical examination, and recovery of hemoglobin to at least 12 g/dL, platelet count to at least 100,000/mm 3 , and granulocyte count to at least 1500/mm 3 . A partial response (PR) required that the percentage of hairy cells in the blood and bone marrow decrease by more than 50%, enlarged organs and lymph nodes decrease by more than 50% by physical examination, and hematologic parameters had to meet the same criteria as for complete response. The table below reports the response rate for 2 groups of patients: (1) Evaluable, ie, patients who could be evaluated for response and (2) Intent-to-Treat, ie, patients diagnosed with hairy cell leukemia.
Parameter FRONTLINE IFN-REFRACTORY a Evaluable
NIPENT
N=138Evaluable
IFN
N=130SWOG 8691 b
Crossover
N=79NCI Phase 2
Studies
N=44Response Rates (%)EvaluableCR84 18 85 58 PR6 24 4 28 Intent-to-TreatN=170 N=170 CR68 14 PR5 18 Median Time to Response (months)CR6.6 11.5 6.0 4.2 PR4.0 6.2 5.8 -- Median Duration of Response (months)CRNR 8.3 NR >7.7 c
(CALGB)
>15.2 c
(MDA)PRNR 15.2 NR -- % Estimated to be in Response After 24 MonthsCR76 16 85 -- PR50 21 -- -- Median Time to Recovery (days)ANC (1500/mm 3 )70 106 -- -- Platelets (100,000/mm 3 )22 36 -- -- NR = Not reached by Kaplan-Meier method; ANC = Absolute neutrophil count. a Evaluable patients b Patients either refractory to, or intolerant of, IFN c Kaplan-Meier estimate The results show that frontline patients treated with NIPENT achieved a significantly higher rate of response than those treated with IFN. The time to recovery of neutrophil and platelet counts was shorter with NIPENT treatment and the estimated duration of response was longer. The response rate in IFN-refractory patients treated with NIPENT was similar to that in NIPENT-treated frontline patients. At a median follow-up duration of 46 months, there was no statistically significant difference in survival between hairy cell leukemia patients initially treated with NIPENT and those initially treated with IFN. However, no definite conclusions regarding survival can be made from these results because they are complicated by the fact that the majority of IFN patients crossed over to NIPENT treatment.
In the Phase 3 SWOG study, 25 patients with hairy cell leukemia died during treatment or follow-up: 18 patients had last received NIPENT (3 of whom had crossed over from IFN), and 7 patients had last received IFN (1 of whom crossed over from NIPENT). Eleven of the 25 deaths occurred within 60 days of the last dose of treatment. Of these, hairy cell leukemia was cited by the investigators as a contributory cause for 1 death in the NIPENT group and 3 deaths in the IFN group. Additionally, infection contributed to the deaths of 3 patients in the NIPENT group and 2 patients in the IFN group. Approximately 4% of hairy cell leukemia patients, in each arm, died more than 60 days after the last dose of either treatment and there was no outstanding cause of death among these patients.
INDICATIONS AND USAGE
NIPENT is indicated as single-agent treatment for both untreated and alpha-interferon-refractory hairy cell leukemia patients with active disease as defined by clinically significant anemia, neutropenia, thrombocytopenia, or disease-related symptoms.
CONTRAINDICATIONS
NIPENT is contraindicated in patients who have demonstrated hypersensitivity to NIPENT.
WARNINGS
See Boxed Warning .
Patients with hairy cell leukemia may experience myelosuppression primarily during the first few courses of treatment. Patients with infections prior to NIPENT treatment have in some cases developed worsening of their condition leading to death, whereas others have achieved complete response. Patients with infection should be treated only when the potential benefit of treatment justifies the potential risk to the patient. Efforts should be made to control the infection before treatment is initiated or resumed.
In patients with progressive hairy cell leukemia, the initial courses of NIPENT treatment were associated with worsening of neutropenia. Therefore, frequent monitoring of complete blood counts during this time is necessary. If severe neutropenia continues beyond the initial cycles, patients should be evaluated for disease status, including a bone marrow examination.
Elevations in liver function tests occurred during treatment with NIPENT and were generally reversible.
Renal toxicity was observed at higher doses in early studies; however, in patients treated at the recommended dose, elevations in serum creatinine were usually minor and reversible. There were some patients who began treatment with normal renal function who had evidence of mild to moderate toxicity at a final assessment. (See DOSAGE AND ADMINISTRATION .)
Rashes, occasionally severe, were commonly reported and may worsen with continued treatment. Withholding of treatment may be required (See DOSAGE AND ADMINISTRATION .)
Acute pulmonary edema and hypotension, leading to death, have been reported in the literature in patients treated with pentostatin in combination with carmustine, etoposide and high dose cyclophosphamide as part of the ablative regimen for bone marrow transplant.
Pregnancy Category D
Pentostatin can cause fetal harm when administered to a pregnant woman. Pentostatin was administered intravenously at doses of 0, 0.01, 0.1, or 0.75 mg/kg/day (0, 0.06, 0.6, and 4.5 mg/m 2 ) to pregnant rats on days 6 through 15 of gestation. Drug-related maternal toxicity occurred at doses of 0.1 and 0.75 mg/kg/day (0.6 and 4.5 mg/m 2 ). Teratogenic effects were observed at 0.75 mg/kg/day (4.5 mg/m 2 ) manifested by increased incidence of various skeletal malformations. In a dose range-finding study, pentostatin was administered intravenously to rats at doses of 0, 0.05, 0.1, 0.5, 0.75, or 1 mg/kg/day (0, 0.3, 0.6, 3, 4.5, 6 mg/m 2 ) on days 6 through 15 of gestation. Fetal malformations that were observed were an omphalocele at 0.05 mg/kg (0.3 mg/m 2 ), gastroschisis at 0.75 mg/kg and 1 mg/kg (4.5 and 6 mg/m 2 ), and a flexure defect of the hindlimbs at 0.75 mg/kg (4.5 mg/m 2 ). Pentostatin was also shown to be teratogenic in mice when administered as a single 2 mg/kg (6 mg/m 2 ) intraperitoneal injection on day 7 of gestation. Pentostatin was not teratogenic in rabbits when administered intravenously on days 6 through 18 of gestation at doses of 0, 0.005, 0.01, or 0.02 mg/kg/day (0, 0.015, 0.03, or 0.06 mg/m 2 ); however, maternal toxicity, abortions, early deliveries, and deaths occurred in all drug-treated groups. There are no adequate and well-controlled studies in pregnant women. If NIPENT is used during pregnancy, or if the patient becomes pregnant while taking (receiving) this drug, the patient should be apprised of the potential hazard to the fetus. Women of childbearing potential receiving NIPENT should be advised to avoid becoming pregnant.
PRECAUTIONS
General
Therapy with NIPENT requires regular patient observation and monitoring of hematologic parameters and blood chemistry values. If severe adverse reactions occur, the drug should be withheld (see DOSAGE AND ADMINISTRATION ), and appropriate corrective measures should be taken according to the clinical judgment of the physician.
NIPENT treatment should be withheld or discontinued in patients showing evidence of nervous system toxicity.
Information for Patients
Patients should be advised of the signs and symptoms of adverse events associated with NIPENT therapy. (See ADVERSE REACTIONS .)
Laboratory Tests
Prior to initiating therapy witn NIPENT, renal function should be assessed with a serum creatinine and/or a creatinine clearance assay. (See CLINICAL PHARMACOLOGY and DOSAGE AND ADMINISTRATION .) Complete blood counts and serum creatinine should be performed before each dose of NIPENT and at other appropriate periods during therapy (see DOSAGE AND ADMINISTRATION ). Severe neutropenia has been observed following the early courses of treatment with NIPENT and therefore frequent monitoring of complete blood counts is recommended during this time. If hematologic parameters do not improve with subsequent courses, patients should be evaluated for disease status, including a bone marrow examination. Periodic monitoring of the peripheral blood for hairy cells should be performed to assess the response to treatment.
In addition, bone marrow aspirates and biopsies may be required at 2 to 3 month intervals to assess the response to treatment.
Drug Interactions
Allopurinol and NIPENT are both associated with skin rashes. Based on clinical studies in 25 refractory patients who received both NIPENT and allopurinol, the combined use of NIPENT and allopurinol did not appear to produce a higher incidence of skin rashes than observed with NIPENT alone. There has been a report of one patient who received both drugs and experienced a hypersensitivity vasculitis that resulted in death. It was unclear whether this adverse event and subsequent death resulted from the drug combination.
Biochemical studies have demonstrated that pentostatin enhances the effects of vidarabine, a purine nucleoside with antiviral activity. The combined use of vidarabine and NIPENT may result in an increase in adverse reactions associated with each drug. The therapeutic benefit of the drug combination has not been established.
The combined use of NIPENT and fludarabine phosphate is not recommended because it may be associated with an increased risk of fatal pulmonary toxicity (see WARNINGS ).
Acute pulmonary edema and hypotension, leading to death, have been reported in the literature in patients treated with pentostatin in combination with carmustine, etoposide and high dose cyclophosphamide as part of the ablative regimen for bone marrow transplant.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis: No animal carcinogenicity studies have been conducted with pentostatin.
Mutagenesis: Pentostatin was nonmutagenic when tested in Salmonella typhimurium strains TA-98, TA-1535, TA-1537, and TA-1538. When tested with strain TA-100, a repeatable statistically significant response trend was observed with and without metabolic activation. The response was 2.1 to 2.2 fold higher than the background at 10 mg/plate, the maximum possible drug concentration. Formulated pentostatin was clastogenic in the in vivo mouse bone marrow micronucleus assay at 20, 120, and 240 mg/kg. Pentostatin was not mutagenic to V79 Chinese hamster lung cells at the HGPRT locus exposed 3 hours to concentrations of 1 to 3 mg/mL, with or without metabolic activation. Pentostatin did not significantly increase chromosomal aberrations in V79 Chinese hamster lung cells exposed 3 hours to 1 to 3 mg/mL in the presence or absence of metabolic activation.
Impairment of Fertility: No fertility studies have been conducted in animals; however, in a 5-day intravenous toxicity study in dogs, mild seminiferous tubular degeneration was observed with doses of 1 and 4 mg/kg. The possible adverse effects on fertility in humans have not been determined.
Pregnancy
Pregnancy Category D: (See WARNINGS )
Nursing Mothers
It is not known whether NIPENT is excreted in human milk. Because many drugs are excreted in human milk, and because of the potential for serious adverse reactions in nursing infants from pentostatin, a decision should be made whether to discontinue nursing or discontinue the drug, taking into account the importance of NIPENT to the mother.
Pediatric Use
Safety and effectiveness in children or adolescents have not been established.
ADVERSE REACTIONS
Most patients treated for hairy cell leukemia in the five NCI-sponsored Phase 2 and the Phase 3 SWOG study experienced an adverse event. The following table lists the most frequently occurring adverse events in patients treated with NIPENT (both frontline and IFN-refractory patients) compared with IFN (frontline only), regardless of drug association. The drug association of some adverse events is uncertain as they may be associated with the disease itself (eg, infection, hematologic suppression), but other events, such as the gastrointestinal symptoms, rashes, and abnormal liver function tests, can in many cases be attributed to the drug. Most adverse events that were asssessed for severity were either mild or moderate, and diminished in frequency with continued therapy.
All Adverse
Events aPercent of Patients Frontline, Treated With
NIPENT
N=180Frontline, Treated With
IFN
N=176IFN-Refractory Treated With
NIPENT
N=197Nausea and/or
Vomiting63 22 53 b Fever46 59 42 Rash43 30 26 Fatigue42 55 29 Leukopenia22 15 60 Pruritus21 6 10 Coughing/Increased
Cough20 15 17 Myalgia19 36 11 Chills19 34 11 Headache17 29 13 Diarrhea17 17 15 Abdominal Pain16 15 4 Anorexia13 10 16 Upper Respiratory
Infection13 8 16 Asthenia12 13 10 Stomatitis12 7 5 Rhinitis11 15 10 Dyspnea11 13 8 Anemia8 5 35 Pain8 19 20 Pharyngitis8 11 10 Sweating Increased/
Sweating8 21 10 Viral Infection8 17 NR Infection7 c 2 c 36 Arthralgia6 14 3 Thrombocytopenia6 6 32 Skin Disorder4 5 17 Allergic Reaction2 1 11 Hepatic Disorder/
Elevated Liver
Function Tests d2 2 19 Neurologic Disorder,
CNS/CNS Toxicity1 NR 11 Lung Disorder/
DiseaseNR 1 12 NauseaNR NR 22 Genitourinary
DisorderNR NR 15 NR = Not Reporteda Occurring in more than 10% of patients, in any group, regardless of drug associationb Includes only nausea with vomitingc These figures represent only unspecified infections. Refer to infection table.d Elevated liver enzymes and liver disorder for SWOGThe total incidence for all types of infections is considerably higher for both treatment groups in the SWOG 8691 study than is listed in the table above. An intent-to-treat analysis of infections found that 38% of patients treated with NIPENT and 34% of patients treated with IFN averaged 2.4 and 1.9 documented infections during treatment, respectively. The following table lists the different types of infections that were reported as adverse events during the initial phase of the SWOG study. There were no apparent differences in the types of infection between the 2 treatment groups, with the possible exception of herpes zoster which was reported more frequently for NIPENT (8%) than for IFN (1%).
Type of Infection
Percent of Patients Frontline,
Treated
With NIPENT
N=180Frontline,
Treated
With IFN
N=176Upper Respiratory Infection 138Rhinitis 1115Herpes Zoster 81Pharyngitis 811Viral Infection 817Infection (Unspecified) 72Sinusitis 64Cellulitis 63Bacterial Infection 54Pneumonia 57Conjunctivitis 42Furunculosis 4<1Herpes Simplex 41Bronchitis 32Sepsis 32Urinary Tract Infection 33Abscess, Skin 24Moniliasis, Oral 2<1Mycotic Infection, Skin <13Osteomyelitis 10The drug relatedness of the adverse events listed below cannot be excluded. The following adverse events occurred in 3% to 10% of NIPENT-treated patients in the initial phase of the SWOG study:
Body as a Whole --Chest Pain, Death, Face Edema, Peripheral Edema
Cardiovascular System --Hemorrhage, Hypotension
Digestive System --Dental Abnormalities, Dyspepsia, Flatulence, Gingivitis
Hemic and Lymphatic System --Agranulocytosis
Laboratory Deviations --Elevated Creatinine
Musculoskeletal System --Arthralgia
Nervous System --Confusion, Dizziness, Insomnia, Paresthesia, Somnolence
Psychobiologic Function --Anxiety, Depression, Nervousness
Respiratory System --Asthma
Skin & Appendages --Skin Dry, Urticaria
The remaining adverse events which occurred in less than 3% of NIPENT-treated patients during the initial phase of the SWOG study:
Body as a Whole --Flu-like Symptoms, Hangover Effect, Neoplasm
Cardiovascular System --Angina Pectoris, Arrhythmia, A-V Block, Bradycardia, Extrasystoles Ventricular, Heart Arrest, Heart Failure, Hypertension, Pericardial Effusion, Phlebitis, Pulmonary Embolus, Sinus Arrest, Tachycardia, Thrombophlebitis Deep, Vasculitis
Digestive System --Constipation, Dysphagia, Glossitis, Ileus
Hemic and Lymphatic System --Acute Leukemia, Anemia-Hemolytic, Aplastic Anemia
Laboratory Deviations --Hypercalcemia, Hyponatremia
Musculoskeletal System --Arthritis, Gout
Nervous System --Amnesia, Ataxia, Convulsions, Dreaming Abnormal, Dysarthria, Encephalitis, Hyperkinesia, Meningism, Neuralgia, Neuritis, Neuropathy, Paralysis, Syncope, Twitching, Vertigo
Psychobiologic Function --Decrease/Loss Libido, Emotional Liability, Hallucination, Hostility, Neurosis, Thinking Abnormal
Respiratory System --Bronchospasm, Larynx Edema
Skin and Appendages --Acne, Alopecia, Eczema, Petechial Rash, Photosensitivity Reaction
Special Senses --Amblyopia, Deafness, Earache, Eyes Dry, Labyrinthitis, Lacrimation Disorder, Nonreactive Eye, Photophobia, Retinopathy, Tinnitus, Unusual Taste, Vision Abnormal, Watery Eyes
Urogenital System --Amenorrhea, Breast Lump, Impotence, Kidney Function Abnormal, Nephropathy, Renal Failure, Renal Insufficiency, Renal Stone
One patient with hairy cell leukemia treated with NIPENT during another clinical study developed unilateral uveitis with vision loss.
Nineteen (5%) patients withdrew from the Phase 3 SWOG 8691 study because of adverse events; 9 during initial NIPENT treatment, 4 during NIPENT crossover, 5 during initial IFN treatment, and 1 during both initial IFN treatment and NIPENT crossover. In the Phase 2 studies in IFN-refractory hairy cell leukemia, 11% of patients withdrew from treatment with NIPENT due to an adverse event.
OVERDOSAGE
No specific antidote for NIPENT overdose is known. NIPENT administered at higher doses (20 to 50 mg/m 2 in divided doses over 5 days) than recommended was associated with deaths due to severe renal, hepatic, pulmonary, and CNS toxicity. In case of overdose, management would include general supportive measures through any period of toxicity that occurs.
DOSAGE AND ADMINISTRATION
It is recommended that patients receive hydration with 500 to 1,000 mL of 5% Dextrose in 0.5 Normal Saline or equivalent before NIPENT administration. An additional 500 mL of 5% Dextrose or equivalent should be administered after NIPENT is given.
The recommended dosage of NIPENT for the treatment of hairy cell leukemia is 4 mg/m 2 every other week. NIPENT may be administered intravenously by bolus injection or diluted in a larger volume and given over 20 to 30 minutes. (See Preparation of Intravenous Solution .)
Higher doses are not recommended.
No extravasation injuries were reported in clinical studies.
The optimal duration of treatment has not been determined. In the absence of major toxicity and with observed continuing improvement, the patient should be treated until a complete response has been achieved. Although not established as required, the administration of two additional doses has been recommended following the achievement of a complete response.
All patients receiving NIPENT at 6 months should be assessed for response to treatment. If the patient has not achieved a complete or partial response, treatment with NIPENT should be discontinued.
If the patient has achieved a partial response, NIPENT treatment should be continued in an effort to achieve a complete response. At any time thereafter that a complete response is achieved, two additional doses of NIPENT are recommended. NIPENT treatment should then be stopped. If the best response to treatment at the end of 12 months is a partial response, it is recommended that treatment with NIPENT be stopped.
Withholding or discontinuation of individual doses may be needed when severe adverse reactions occur. Drug treatment should be withheld in patients with severe rash, and withheld or discontinued in patients showing evidence of nervous system toxicity.
NIPENT treatment should be withheld in patients with active infection occurring during the treatment but may be resumed when the infection is controlled.
Patients who have elevated serum creatinine should have their dose withheld and a creatinine clearance determined. There are insufficient data to recommend a starting or a subsequent dose for patients with impaired renal function (creatinine clearance <60 mL/min).
Patients with impaired renal function should be treated only when the potential benefit justifies the potential risk. Two patients with impaired renal function (creatinine clearances 50 to 60 mL/min) achieved complete response without unusual adverse events when treated with 2 mg/m 2 .
No dosage reduction is recommended at the start of therapy with NIPENT in patients with anemia, neutropenia, or thrombocytopenia. In addition, dosage reductions are not recommended during treatment in patients with anemia and thrombocytopenia if patients can be otherwise supported hematologically. NIPENT should be temporarily withheld if the absolute neutrophil count falls during treatment below 200 cells/mm 3 in a patient who had an initial neutrophil count greater than 500 cells/mm 3 and may be resumed when the count returns to predose levels.
Preparation of Intravenous Solution
- Procedures for proper handling and disposal of anticancer drugs should be followed. Several guidelines on this subject have been published. 2-7 There is no general agreement that all of the procedures recommended in the guidelines are necessary or appropriate. Spills and wastes should be treated with a 5% sodium hypochlorite solution prior to disposal.
- Protective clothing including polyethylene gloves must be worn.
- Transfer 5 mL of Sterile Water for Injection, USP to the vial containing NIPENT and mix thoroughly to obtain complete dissolution of a solution yielding 2 mg/mL. Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration.
- NIPENT may be given intravenously by bolus injection or diluted in a larger volume (25 to 50 mL) with 5% Dextrose Injection, USP or 0.9% Sodium Chloride Injection, USP. Dilution of the entire contents of a reconstituted vial with 25 mL or 50 mL provides a pentostatin concentration of 0.33 mg/mL or 0.18 mg/mL, respectively, for the diluted solutions.
- NIPENT solution when diluted for infusion with 5% Dextrose Injection, USP or 0.9% Sodium Chloride Injection, USP does not interact with PVC infusion containers or administration sets at concentrations of 0.18 mg/mL to 0.33 mg/mL.
Stability
NIPENT vials are stable at refrigerated storage temperature 2° to 8°C (36° to 46°F) for the period stated on the package. Vials reconstituted or reconstituted and further diluted as directed may be stored at room temperature and ambient light but should be used within 8 hours because NIPENT contains no preservatives.
HOW SUPPLIED
NIPENT (pentostatin for injection) is supplied as a sterile lyophilized white to off-white powder in single-dose vials containing 10 mg of pentostatin. The vials are packed in individual cartons. NDC 62701-800-01
Storage: Store NIPENT vials under refrigerated storage conditions 2° to 8°C (36° to 46°F).
Rx Only
REFERENCES
- Malspeis L, et al. Clinical Pharmacokinetics of 2'-Deoxycoformycin. Cancer Treatment Symposia 2:7-15, 1984.
- Recommendations for the safe handling of parenteral antineoplastic drugs. NIH publication 83-2621. For sale by the Superintendent of Documents, US Government Printing Office, Washington, NC 20402.
- AMA council report. Guidelines for handling parenteral antineoplastics. JAMA 253:1590-2, 1985.
- National Study Commission on Cytotoxic Exposure-Recommendations for handling cytotoxic agents. Available from Louis P. Jeffery, Sc.D., Chairman, National Study Commission on Cytotoxic Exposure, Massachusetts College of Pharmacy and Allied Health Sciences, 179 Longwood Ave, Boston, Massachusetts 02115.
- Clinical Oncology Society of Australia: Guidelines and recommendations for safe handling of antineoplastic agents. Med J Australia 1:426-8, 1983.
- Jones RB, et al. Safe handling of chemotherapeutic agents: A report from the Mount Sinai Medical Center. CA: A Cancer Journal for Clinicians 33:258-63, 1983.
-
American Society of Hospital Pharmacists technical assistance bulletin on handling cytotoxic and hazardous drugs. Am J Hosp Pharm 47:1033-49, 1990.
800P1 Rev. April, 1998
Subscribe to the "News" RSS Feed 
TOP ۞