-
Zemplar Injection (Abbott)
DESCRIPTION
Zemplar ® (paricalcitol injection, USP) is a synthetically manufactured vitamin D analog. It is available as a sterile, clear, colorless, aqueous solution for intravenous injection. Each mL contains paricalcitol, USP, 5 mcg; propylene glycol, 30% (v/v); and alcohol, 20% (v/v).
Paricalcitol is a white powder chemically designated as 19-nor-1(alpha),3(beta),25-trihydroxy-9,10-secoergosta-5(Z),7(E),22(E)-triene and has the following structural formula:
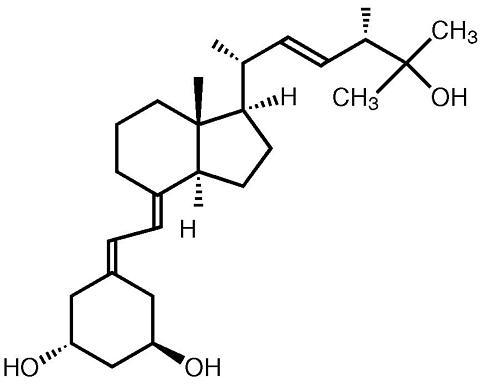
Molecular formula is C 27 H 44 O 3 .
Molecular weight is 416.65.
CLINICAL PHARMACOLOGY
Mechanism of Action
Paricalcitol is a synthetic vitamin D analog. Vitamin D and paricalcitol have been shown to reduce parathyroid hormone (PTH) levels.
Pharmacokinetics
Distribution
The pharmacokinetics of paricalcitol have been studied in patients with chronic renal failure (CRF) requiring hemodialysis. Zemplar ® is administered as an intravenous bolus injection. Within two hours after administering doses ranging from 0.04 to 0.24 mcg/kg, concentrations of paricalcitol decreased rapidly; thereafter, concentrations of paricalcitol declined log-linearly with a mean half-life of about 15 hours. No accumulation of paricalcitol was observed with multiple dosing.
Elimination
In healthy subjects, plasma radioactivity after a single 0.16 mcg/kg intravenous bolus dose of 3 H-paricalcitol (n=4) was attributed to parent drug. Paricalcitol was eliminated primarily by hepatobiliary excretion, as 74% of the radioactive dose was recovered in feces and only 16% was found in urine.
Metabolism
Several unknown metabolites were detected in both the urine and feces, with no detectable paricalcitol in the urine. These metabolites have not been characterized and have not been identified. Together, these metabolites contributed 51% of the urinary radioactivity and 59% of the fecal radioactivity. In vitro plasma protein binding of paricalcitol was extensive (>99.9%) and nonsaturable over the concentration range of 1 to 100 ng/mL.
Paricalcitol Pharmacokinetic Characteristics in
CRF Patients (0.24 mcg/kg dose)ParameternValues (Mean ± SD)C max (5 min. after bolus)61850 ± 664 (pg/mL)AUC 0-(infinity)527382 ± 8230 (pg·hr/mL)CL50.72 ± 0.24 (L/hr)V ss56 ± 2 (L)Laboratory Tests
In placebo-controlled studies, paricalcitol reduced serum total alkaline phosphatase levels.
Special Populations
Paricalcitol pharmacokinetics have not been investigated in geriatric or pediatric populations, or for drug-drug interactions. Pharmacokinetics were not gender-dependent.
The disposition of paricalcitol was compared in patients with mild (n=5) and moderate (n=5) hepatic impairment (as indicated by the Child-Pugh method) and subjects with normal hepatic function (n=10). Following administration of a single dose, the pharmacokinetics of unbound paricalcitol were similar across hepatic function groups. Paricalcitol binding to plasma proteins was very high in all hepatic function groups (mean values >99.7%). The protein binding of paricalcitol was decreased in subjects with moderate (but not mild) hepatic impairment; total paricalcitol concentrations tended to be lower for subjects with moderate hepatic impairment compared to the other two hepatic function groups. No dosing adjustment is required in patients with mild and moderate hepatic impairment. The influence of severe hepatic impairment on the pharmacokinetics of paricalcitol has not been evaluated.
Clinical Studies
In three 12-week, placebo-controlled, phase 3 studies in chronic renal failure patients on dialysis, the dose of Zemplar ® was started at 0.04 mcg/kg 3 times per week. The dose was increased by 0.04 mcg/kg every 2 weeks until intact parathyroid hormone (iPTH) levels were decreased at least 30% from baseline or a fifth escalation brought the dose to 0.24 mcg/kg, or iPTH fell to less than 100 pg/mL, or the Ca × P product was greater than 75 within any 2 week period, or serum calcium became greater than 11.5 mg/dL at any time.
Patients treated with Zemplar ® achieved a mean iPTH reduction of 30% within 6 weeks. In these studies, there was no significant difference in the incidence of hypercalcemia or hyperphosphatemia between Zemplar ® and placebo-treated patients. The results from these studies are as follows:
Group
(No. of Pts.)Baseline Mean
(Range)Mean (SE) Change
From Baseline to
Final EvaluationPTH (pg/mL)Zemplar ® (n=40) 783 (291-2076) -379 (43.7) placebo (n=38) 745 (320-1671) -69.6 (44.8) Alkaline Phosphatase (U/L)Zemplar ® (n=31) 150 (40-600) -41.5 (10.6) placebo (n=34) 169 (56-911) +2.6 (10.1) Calcium (mg/dL)Zemplar ® (n=40) 9.3 (7.2-10.4) +0.47 (0.1) placebo (n=38) 9.1 (7.8-10.7) +0.02 (0.1) Phosphorus (mg/dL)Zemplar ® (n=40) 5.8 (3.7-10.2) +0.47 (0.3) placebo (n=38) 6.0 (2.8-8.8) -0.47 (0.3) Calcium ×
Phosphorus ProductZemplar ® (n=40) 54 (32-106) +7.9 (2.2) placebo (n=38) 54 (26-77) -3.9 (2.3)
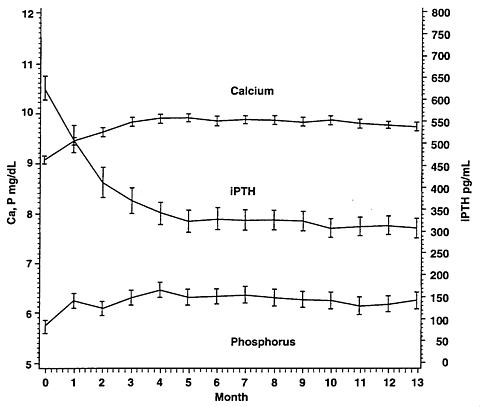
A long-term, open-label safety study of 164 CRF patients (mean dose of 7.5 mcg three times per week), demonstrated that mean serum Ca, P, and Ca × P remained within clinically appropriate ranges with PTH reduction (mean decrease of 319 pg/mL at 13 months).
INDICATIONS AND USAGE
Zemplar ® is indicated for the prevention and treatment of secondary hyperparathyroidism associated with chronic renal failure. Studies in patients with chronic renal failure show that Zemplar ® suppresses PTH levels with no significant difference in the incidence of hypercalcemia or hyperphosphatemia when compared to placebo. However, the serum phosphorus, calcium and calcium × phosphorus product (Ca × P) may increase when Zemplar ® is administered.
CONTRAINDICATIONS
Zemplar ® should not be given to patients with evidence of vitamin D toxicity, hypercalcemia, or hypersensitivity to any ingredient in this product (see PRECAUTIONS , General ).
WARNINGS
Acute overdose of Zemplar ® may cause hypercalcemia, and require emergency attention. During dose adjustment, serum calcium and phosphorus levels should be monitored closely (e.g., twice weekly). If clinically significant hypercalcemia develops, the dose should be reduced or interrupted. Chronic administration of Zemplar ® may place patients at risk of hypercalcemia, elevated Ca × P product, and metastatic calcification. Signs and symptoms of vitamin D intoxication associated with hypercalcemia include:
Early
Weakness, headache, somnolence, nausea, vomiting, dry mouth, constipation, muscle pain, bone pain, and metallic taste.
Late
Anorexia, weight loss, conjunctivitis (calcific), pancreatitis, photophobia, rhinorrhea, pruritus, hyperthermia, decreased libido, elevated BUN, hypercholesterolemia, elevated AST and ALT, ectopic calcification, hypertension, cardiac arrhythmias, somnolence, death, and rarely, overt psychosis.
Treatment of patients with clinically significant hypercalcemia consists of immediate dose reduction or interruption of Zemplar ® therapy and includes a low calcium diet, withdrawal of calcium supplements, patient mobilization, attention to fluid and electrolyte imbalances, assessment of electrocardiographic abnormalities (critical in patients receiving digitalis), and hemodialysis or peritoneal dialysis against a calcium-free dialysate, as warranted. Serum calcium levels should be monitored frequently until normocalcemia ensues.
Phosphate or vitamin D-related compounds should not be taken concomitantly with Zemplar ® .
PRECAUTIONS
General: Digitalis toxicity is potentiated by hypercalcemia of any cause, so caution should be applied when digitalis compounds are prescribed concomitantly with Zemplar ® . Adynamic bone lesions may develop if PTH levels are suppressed to abnormal levels.
Information for the Patient: The patient should be instructed that, to ensure effectiveness of Zemplar ® therapy, it is important to adhere to a dietary regimen of calcium supplementation and phosphorus restriction. Appropriate types of phosphate-binding compounds may be needed to control serum phosphorus levels in patients with chronic renal failure (CRF), but excessive use of aluminum containing compounds should be avoided. Patients should also be carefully informed about the symptoms of elevated calcium
Essential Laboratory Tests: During the initial phase of medication, serum calcium and phosphorus should be determined frequently (e.g., twice weekly). Once dosage has been established, serum calcium and phosphorus should be measured at least monthly. Measurements of serum or plasma PTH are recommended every 3 months. An intact PTH (iPTH) assay is recommended for reliable detection of biologically active PTH in patients with CRF. During dose adjustment of Zemplar ® , laboratory tests may be required more frequently.
Drug Interactions: Specific interaction studies were not performed. Digitalis toxicity is potentiated by hypercalcemia of any cause, so caution should be applied when digitalis compounds are prescribed concomitantly with Zemplar ® .
Carcinogenesis, Mutagenesis, Impairment of Fertility: In a 104-week carcinogenicity study in CD-1 mice, an increased incidence of uterine leiomyoma and leiomyosarcoma was observed at subcutaneous doses of 1 to 10 mcg/kg (< 1 to 3 times the maximum recommended human weekly dose of 0.72 mcg/kg, based on body surface area, mg/m 2 ). The incidence rate of uterine leiomyoma was significantly different than the control group at the highest dose of 10 mcg/kg.
In a 104-week carcinogenicity study in rats, there was an increased incidence of benign adrenal pheochromocytoma at subcutaneous doses of 0.15 to 1.5 mcg/kg (</= 1 times the maximum recommended human weekly dose of 0.72 mcg/kg, based on body surface area, mg/m 2 ). The increased incidence of pheochromocytomas in rats may be related to the alteration of calcium homeostasis by paricalcitol. In carcinogenicity studies in rats and mice, paricalcitol did not affect the incidences of tumors apart from benign rodent-specific lesions related to the effects of chronic hypercalcemia.
Paricalcitol did not exhibit genetic toxicity in vitro with or without metabolic activation in the microbial mutagenesis assay (Ames Assay), mouse lymphoma mutagenesis assay (L5178Y), or a human lymphocyte cell chromosomal aberration assay. There was also no evidence of genetic toxicity in an in vivo mouse micronucleus assay. Zemplar ® had no effect on fertility (male or female) in rats at intravenous doses up to 20 mcg/kg/dose [equivalent to 13 times the highest recommended human dose (0.24 mcg/kg) based on surface area, mg/m 2 ].
Pregnancy: Pregnancy Category C. Paricalcitol has been shown to cause minimal decreases in fetal viability (5%) when administered daily to rabbits at a dose 0.5 times the 0.24 mcg/kg human dose (based on surface area, mg/m 2 ) and when administered to rats at a dose 2 times the 0.24 mcg/kg human dose (based on plasma levels of exposure). At the highest dose tested (20 mcg/kg 3 times per week in rats, 13 times the 0.24 mcg/kg human dose based on surface area), there was a significant increase of the mortality of newborn rats at doses that were maternally toxic (hypercalcemia). No other effects on offspring development were observed. Paricalcitol was not teratogenic at the doses tested.
There are no adequate and well-controlled studies in pregnant women. Zemplar ® should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Nursing Mothers: It is not known whether paricalcitol is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when Zemplar ® is administered to a nursing woman.
Pediatric Use: The safety and effectiveness of Zemplar ® were examined in a 12-week randomized, double-blind, placebo-controlled study of 29 pediatric patients, aged 5-19 years, with end-stage renal disease on hemodialysis and nearly all had received some form of vitamin D prior to the study. Seventy-six percent of the patients were male, 52% were Caucasian and 45% were African-American. The initial dose of Zemplar ® was 0.04 mcg/kg 3 times per week based on baseline iPTH level of less than 500 pg/mL, or 0.08 mcg/kg 3 times a week based on baseline iPTH level of >/=500pg/mL, respectively. The dose of Zemplar ® was adjusted in 0.04 mcg/kg increments based on the levels of serum iPTH, calcium and Ca × P. The mean baseline levels of iPTH were 841 pg/mL for the 15 Zemplar ® treated patients and 740 pg/mL for the 14 placebo-treated subjects. The mean dose of Zemplar ® administered was 4.6 mcg (range: 0.8 mcg-9.6 mcg). Ten of the 15 (67%) Zemplar-treated patients and 2 of the 14 (14%) placebo-treated subjects completed the trial. Ten of the placebo patients (71%) were discontinued due to excessive elevations in iPTH levels as defined by 2 consecutive iPTH levels >700 pg/mL and greater than baseline after 4 weeks of treatment.
In the primary efficacy analysis, 9 of 15 (60%) subjects in the Zemplar ® group had 2 consecutive 30% decreases from baseline iPTH compared with 3 of 14 (21%) patients in the placebo group (95% CI for the difference between groups -1%, 63%). Twenty-three percent of Zemplar ® vs. 31% of placebo patients had at least one serum calcium level >10.3 mg/dL and 40% vs. 14% of Zemplar ® vs. placebo subjects had at least one Ca × P ion product >72 (mg/dL) 2 . The overall percentage of serum calcium measurements >10.3 mg/dL was 7% in the Zemplar ® group and 7% in the placebo group; the overall percentage of patients with Ca × P product > 72 (mg/dL) 2 was 8% in the Zemplar ® group and 7% in the placebo group. No subjects in either the Zemplar ® group or placebo group developed hypercalcemia (defined as at least one calcium value >11.2 mg/dL) during the study.
Geriatric Use: Of the 40 patients receiving Zemplar ® in the three phase 3 placebo-controlled CRF studies, 10 patients were 65 years or over. In these studies, no overall differences in efficacy or safety were observed between patients 65 years or older and younger patients.
ADVERSE REACTIONS
Zemplar ® has been evaluated for safety in clinical studies in 454 CRF patients. In four, placebo-controlled, double-blind, multicenter studies, discontinuation of therapy due to any adverse event occurred in 6.5% of 62 patients treated with Zemplar ® (dosage titrated as tolerated, see CLINICAL PHARMACOLOGY , Clinical Studies ) and 2.0% of 51 patients treated with placebo for one to three months. Adverse events occurring with greater frequency in the Zemplar ® group at a frequency of 2% or greater, regardless of causality, are presented in the following table:
Adverse Event Incidence Rates for All Treated Patients
In All Placebo-Controlled StudiesAdverse EventZemplar ®
(n=62)%Placebo
(n=51)%Overall71 78 Body as a WholeChills5 0 Feeling unwell3 0 Fever5 2 Flu5 4 Sepsis5 2 Cardiovascular SystemPalpitation3 0 Digestive SystemDry mouth3 2 Gastrointestinal
bleeding5 2 Nausea13 8 Vomiting8 4 Metabolic and Nutritional DisordersEdema7 0 Nervous SystemLight-headedness5 2 Respiratory SystemPneumonia5 0
A patient who reported the same medical term more than once was counted only once for that medical term.
Safety parameters (changes in mean Ca, P, Ca × P) in an open-label safety study up to 13 months in duration support the long-term safety of Zemplar ® in this patient population.
Adverse Events during post-marketing experience: Taste perversion, such as metallic taste, and allergic reactions, such as rash, urticaria and pruritus rarely have been reported.
OVERDOSAGE
Overdosage of Zemplar ® may lead to hypercalcemia (see WARNINGS ).
DOSAGE AND ADMINISTRATION
The currently accepted target range for iPTH levels in CRF patients is no more than 1.5 to 3 times the non-uremic upper limit of normal.
The recommended initial dose of Zemplar ® is 0.04 mcg/kg to 0.1 mcg/kg (2.8 - 7 mcg) administered as a bolus dose no more frequently than every other day at any time during dialysis. Doses as high as 0.24 mcg/kg (16.8 mcg) have been safely administered.
If a satisfactory response is not observed, the dose may be increased by 2 to 4 mcg at 2- to 4-week intervals. During any dose adjustment period, serum calcium and phosphorus levels should be monitored more frequently, and if an elevated calcium level or a Ca × P product greater than 75 is noted, the drug dosage should be immediately reduced or interrupted until these parameters are normalized. Then, Zemplar ® should be reinitiated at a lower dose. Doses may need to be decreased as the PTH levels decrease in response to therapy. Thus, incremental dosing must be individualized.
The following table is a suggested approach in dose titration:
Suggested Dosing Guidelines PTH LevelZemplar ® Dosethe same or increasingincreasedecreasing by <30%increasedecreasing by >30%, <60%maintaindecreasing by >60%decreaseone and one-half to three
times upper limit of normalmaintain
The influence of mild to moderately impaired hepatic function on paricalcitol pharmacokinetics is sufficiently small that no dosing adjustment is required.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration whenever solution and container permit.
Discard unused portion.
HOW SUPPLIED
Zemplar ® (paricalcitol injection, USP) 5 mcg/mL is supplied as 1 and 2 mL single-dose Fliptop Vials.
List No. Volume/Container Concentration Total Content 1658 1 mL/Fliptop Vial 5 mcg/mL 5 mcg 1658 2 mL/Fliptop Vial 5 mcg/mL 10 mcg
Store at 25°C (77°F). Excursions permitted to 15°-30°C (59°-86°F).
U.S. patents: 5,246,925; 5,587,497
Ref.: EN-0423 (10/04)
Revised: October, 2004
©Abbott 2004
Manufactured by
Hospira, Inc.
Lake Forest, IL 60045 USA
For
Abbott Laboratories
North Chicago, IL 60064 USA
Subscribe to the "News" RSS Feed 
TOP ۞